Loading metrics
Open Access
Pearls provide concise, practical and educational insights into topics that span the pathogens field.
See all article types »

Emerging Infectious Diseases: Threats to Human Health and Global Stability
* E-mail: [email protected]
Affiliation National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, United States of America
- David M. Morens,
- Anthony S. Fauci
Published: July 4, 2013
- https://doi.org/10.1371/journal.ppat.1003467
- Reader Comments
Citation: Morens DM, Fauci AS (2013) Emerging Infectious Diseases: Threats to Human Health and Global Stability. PLoS Pathog 9(7): e1003467. https://doi.org/10.1371/journal.ppat.1003467
Editor: Joseph Heitman, Duke University Medical Center, United States of America
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication.
Funding: Both authors are employees of NIH and as such our work is funded by NIH from operating funds rather than grants or other awards.
Competing interests: The authors have declared that no competing interests exist.
The inevitable, but unpredictable, appearance of new infectious diseases has been recognized for millennia, well before the discovery of causative infectious agents. Today, however, despite extraordinary advances in development of countermeasures (diagnostics, therapeutics, and vaccines), the ease of world travel and increased global interdependence have added layers of complexity to containing these infectious diseases that affect not only the health but the economic stability of societies. HIV/AIDS, severe acute respiratory syndrome (SARS), and the most recent 2009 pandemic H1N1 influenza are only a few of many examples of emerging infectious diseases in the modern world [1] ; each of these diseases has caused global societal and economic impact related to unexpected illnesses and deaths, as well as interference with travel, business, and many normal life activities. Other emerging infections are less catastrophic than these examples; however, they nonetheless may take a significant human toll as well as cause public fear, economic loss, and other adverse outcomes.
Determinants of Emergence and Reemergence
Historical information as well as microbial sequencing and phylogenetic constructions make it clear that infectious diseases have been emerging and reemerging over millennia, and that such emergences are driven by numerous factors ( Table 1 ). Notably, 60 to 80 percent of new human infections likely originated in animals, disproportionately rodents and bats, as shown by the examples of hantavirus pulmonary syndrome, Lassa fever, and Nipah virus encephalitis [2] – [4] . Most other emerging/reemerging diseases result from human-adapted infectious agents that genetically acquire heightened transmission and/or pathogenic characteristics. Examples of such diseases include multidrug-resistant and extensively drug-resistant (MDR and XDR) tuberculosis, toxin-producing Staphylococcus aureus causing toxic shock syndrome, and pandemic influenza [1] – [10] .
- PPT PowerPoint slide
- PNG larger image
- TIFF original image
https://doi.org/10.1371/journal.ppat.1003467.t001
Although precise figures are lacking, emerging infectious diseases comprise a substantial fraction of all consequential human infections. They have caused the deadliest pandemics in recorded human history, including the Black Death pandemic (bubonic/pneumonic plague; 25–40 million deaths) in the fourteenth century, the 1918 influenza pandemic (50 million deaths), and the HIV/AIDS pandemic (35 million deaths so far) [6] , [9] .
Definition and Concepts
Two major categories of emerging infections— newly emerging and reemerging infectious diseases—can be defined, respectively, as diseases that are recognized in the human host for the first time; and diseases that historically have infected humans, but continue to appear in new locations or in drug-resistant forms, or that reappear after apparent control or elimination [1] . Emerging/reemerging infections may exhibit successive stages of emergence. These stages include adaptation to a new host [11] , an epidemic/pathogenic stage, an endemic stage, and a fully adapted stage in which the organism may become nonpathogenic and potentially even beneficial to the new host (e.g., the human gut microbiome) or stably integrated into the host genome (e.g., as endogenous retroviruses). Although these successive stages characterize the evolution of certain microbial agents more than others, they nevertheless can provide a useful framework for understanding many of the dynamic relationships between microorganisms, human hosts, and the environment.
It is also worth noting that the dynamic and complicated nature of many emerging infections often leaves distinctions between emerging and reemerging infections open to question, leading various experts to classify them differently. For example, we describe as “reemerging” new or more severe diseases associated with acquisition of new genes by an existing microbe, e.g., antibiotic resistance genes, even when mutations cause entirely new diseases with unique clinical epidemiologic features, e.g., Brazilian purpuric fever [12] . Similarly, we refer to SARS as an emerging disease a decade after it disappeared, and apply the same term to the related MERS (Middle East Respiratory Syndrome) β coronavirus which appeared in Saudi Arabia in late 2012 [13] .
Examples of Newly Emerging Infectious Diseases
The most salient modern example of an emerging infectious disease is HIV/AIDS, which likely emerged a century ago after multiple independent events in which the virus jumped from one primate host to another (chimpanzees to humans) and subsequently, as a result of a complex array of social and demographic factors, spread readily within the human population. AIDS was not recognized as a distinct entity until 1981 [6] , [9] , after its initial detection among certain risk groups, such as men who have sex with men, recipients of blood products, and injection drug users. It was soon apparent, however, that the disease was not restricted to these groups, and indeed, the bulk of HIV infections globally has resulted from heterosexual transmission that has been heavily weighted within the developing world, particularly sub-Saharan Africa where a number of factors were responsible for this rapid spread; chief among these were human movement along truck routes accompanied by a high level of commercial sex work, inadequate public health infrastructures, poverty, and social inequality.
Other examples of disease emergences [1] – [10] include SARS, which emerged from bats and spread into humans first by person-to-person transmission in confined spaces, then within hospitals, and finally by human movement between international air hubs. Nipah virus also emerged from bats and caused an epizootic in herds of intensively bred pigs, which in turn served as the animal reservoir from which the virus was passed on to humans. The 2009 H1N1 pandemic influenza virus emerged from pigs as well, but only after complex exchanges of human, swine, and avian influenza genes [14] . H5N1 influenza emerged from wild birds to cause epizootics that amplified virus transmission in domestic poultry, precipitating dead-end viral transmission to poultry-exposed humans. Additional examples are many [1] – [10] ; however, the variables associated with emergences are unique for each and typically complex.
Examples of Reemerging Infectious Diseases
Most of the important reemerging infectious disease agents first appeared long ago, but have survived and persisted by adapting to changing human populations and to environments that have been altered by humans. Dengue virus and West Nile virus (WNV), distantly related flaviviruses, serve as good examples. They have been spread by geographic movement of humans in association with the mosquito vectors for the diseases. For example, dengue came to the Americas in association with the slave trade of earlier centuries. In this regard, slaves infected by mosquitoes in Africa presumably brought the infection to the Americas by seeding the mosquito population upon arrival [15] . Similarly, WNV came to the United States in 1999 when an infected human, bird, or mosquito came by air travel from the Middle East to the Western Hemisphere, providing a source for introduction of infection to New World mosquitoes and birds. Pathogenic strains of dengue have also spread back from Southeast Asia to the Western Hemisphere, as has a major mosquito vector, Aedes albopictus . Unlike most arboviruses, which are partly or completely host-restricted, WNV has become adapted to multiple mosquito and avian species, a major factor in increasing its opportunity to infect humans. The lack of additional hosts undoubtedly drove the mosquitoes that are the vectors of dengue and the dengue virus itself to favor adapting to humans and to their behaviors and environments. The association of dengue with Aedes mosquitoes that live in and around human habitations mean that crowding, poor sanitation, and poverty provide ideal environments for transmission to humans [15] . Host immunity factors are also thought to be involved in the severe/fatal form of dengue known as dengue shock syndrome [15] .
Other non-arboviral examples of emerging infections abound. For example, cholera has repeatedly reemerged over more than two centuries in association with global travel, changing seasons, war, natural disasters, and conditions that lead to inadequate sanitation, poverty, and social disruption. Emergences of disease caused by community- and hospital-acquired Clostridium difficile and methicillin-resistant Staphylococcus aureus (MRSA) have been driven by increased and/or inappropriate use of antibiotics, and some hospital-acquired organisms such as MRSA have now moved into community transmission. The global emergence of plasmid-spread NDM-1 (New Delhi β-lactamase) Gram-negative pan-resistant organisms, linked to global antibiotic use and inadequate antibiotic stewardship, medical tourism, economic globalization, and other aspects of modern life, has prompted calls for development of international control mechanisms [16] that are applicable to a number of emerging bacterial diseases in the developing and developed world. Drug resistance mutations have also caused the reemergences of certain pathogens such as multidrug-resistant and extensively drug-resistant tuberculosis, drug-resistant malaria, and numerous bacterial diseases such as vancomycin-resistant enterococci. Fungi have made significant contributions to disease emergence as well. In Africa, cryptococcal disease has already surpassed tuberculosis as a leading cause of death [17] . Other examples of fungal emergence include comorbidities in HIV-infected individuals (17), Cryptococcus gattii epidemics in predominantly healthy persons in the U.S. [18] , [19] , and a 2012 U.S. nationwide epidemic of Exserohilum rostratum infections associated with contaminated pharmaceutical products [20] .
Will We Ever Eliminate Emerging Infectious Diseases?
While it has become possible to eradicate certain infectious diseases (smallpox and the veterinary disease rinderpest), and to significantly control many others (dracunculiasis, measles, and polio, among others), it seems unlikely that we will eliminate most emerging infectious diseases in the foreseeable future. Pathogenic microorganisms can undergo rapid genetic changes, leading to new phenotypic properties that take advantage of changing host and environmental opportunities. Influenza viruses serve as a good example of emerging and reemerging infectious agents in their ability to rapidly evolve in response to changing host and environmental circumstances via multiple genetic mechanisms. New “founder” influenza viruses [21] appear periodically, cause a pandemic, raise widespread population immunity, and then, in response to human immune pressures, evolve and persist for decades using multiple genetic evolutionary mechanisms to sustain continual immune escape. The 1918 influenza pandemic virus is one example: over the past 95 years, its descendants have evolved continually by antigenic drift, intra-subtypic reassortment, and antigenic shift, the latter producing new pandemics in 1957 and 1968 [14] . Even the genetically complex 2009 pandemic H1N1 influenza virus is a descendant of the 1918 virus [14] . Such continuous genetic hyper-evolution forces us to develop new influenza vaccines containing new antigens on an annual basis.
In the meantime, new human diseases keep emerging. As noted, in late 2012 the novel MERS coronavirus emerged in Saudi Arabia [13] , and in early 2013 a new H7N9 avian influenza virus became epizootic in Eastern China, causing 132 spillover infections of humans (as of June 7, 2013), with 28 percent case fatality [10] , [22] . Its pandemic potential, if any, remains to be determined. Whether or not such outbreaks become more widespread, they nonetheless attract global attention and require significant international effort to monitor and contain. Microbial advantages can be met and overcome only by aggressive vigilance, ongoing dedicated research, and rapid development and deployment of such countermeasures as surveillance tools, diagnostics, drugs, and vaccines.
We appear to be entering a new era in which several important emerging, reemerging, and stable infectious diseases are becoming better controlled (e.g., hepatitis B, rabies, Haemophilus influenzae type B, and even to some extent HIV/AIDS). However, our success in stopping the many new emerging diseases that will inevitably appear is not assured. We have many tools in our armamentarium, including preparedness plans and stockpiles of drugs and vaccines. But each new disease brings unique challenges, forcing us to continually adapt to ever-shifting threats [1] – [10] , [23] . The battle against emerging infectious diseases is a continual process; winning does not mean stamping out every last disease, but rather getting out ahead of the next one.
- View Article
- Google Scholar
- 2. Committee on Microbial Threats to Health, Institute of Medicine (1992) Emerging infections: microbial threats to health in the United States. Washington, DC: National Academy Press.
- 10. World Health Organization. Global Alert and Response (GAR). Disease Outbreak News. Available: http://www.who.int/csr/don/en/index.html . Accessed 5 June 2013.
Login to your account
If you don't remember your password, you can reset it by entering your email address and clicking the Reset Password button. You will then receive an email that contains a secure link for resetting your password
If the address matches a valid account an email will be sent to __email__ with instructions for resetting your password
| Property | Value |
|---|---|
| Status | |
| Version | |
| Ad File | |
| Disable Ads Flag | |
| Environment | |
| Moat Init | |
| Moat Ready | |
| Contextual Ready | |
| Contextual URL | |
| Contextual Initial Segments | |
| Contextual Used Segments | |
| AdUnit | |
| SubAdUnit | |
| Custom Targeting | |
| Ad Events | |
| Invalid Ad Sizes |
Access provided by
Emerging Pandemic Diseases: How We Got to COVID-19
- Editorial The Strength of Curiosity and Purpose Cell November 12, 2020
Download started
- Download PDF Download PDF
- disease ecology
- medical history
Introduction
Infectious diseases that have emerged in the past.
| Year | Name | Deaths | Comments |
|---|---|---|---|
| 430 BCE | “Plague of Athens” | ∼100,000 | First identified trans-regional pandemic |
| 541 | Justinian plague ( ) | 30–50 million | Pandemic; killed half of world population |
| 1340s | “Black Death” ( ) | ∼50 million | Pandemic; killed at least a quarter of world population |
| 1494 | Syphilis ( ) | >50,000 | Pandemic brought to Europe from the Americas |
| c. 1500 | Tuberculosis | High millions | Ancient disease; became pandemic in Middle Ages |
| 1520 | ( ) | 3.5 million | Pandemic brought to New World by Europeans |
| 1793–1798 | “The American plague” | ∼25,000 | Yellow fever terrorized colonial America |
| 1832 | 2nd cholera pandemic (Paris) | 18,402 | Spread from India to Europe/Western Hemisphere |
| 1918 | “Spanish” influenza | ∼50 million | Led to additional pandemics in 1957, 1968, 2009 |
| 1976–2020 | Ebola | 15,258 | First recognized in 1976; 29 regional epidemics to 2020 |
| 1981 | Acute hemorrhagic conjunctivitis | rare deaths | First recognized in 1969; pandemic in 1981 |
| 1981 | HIV/AIDS | ∼37 million | First recognized 1981; ongoing pandemic |
| 2002 | SARS | 813 | Near-pandemic |
| 2009 | H1N1 “swine flu” | 284,000 | 5th influenza pandemic of century |
| 2014 | Chikungunya | uncommon | Pandemic, mosquito-borne |
| 2015 | Zika | ∼1,000? | Pandemic, mosquito-borne |
- Open table in a new tab

Definitions of Emerging Infectious Diseases

| Newly emerging infectious diseases | Diseases recognized in humans for the first time, e.g., HIV/AIDS (1981), Nipah virus (1999), SARS (2002), MERS (2012), COVID-19 (2019) |
| Re-emerging infectious diseases | Diseases that have historically infected humans but continue to re-appear either in new locations (e.g., West Nile in the United States and Russia in 1999) or in resistant forms (e.g., methicillin-resistant ) |
| Deliberately emerging infectious diseases | Diseases associated with intent to harm, including mass bioterrorism |
| Accidentally emerging infectious diseases | Diseases created by humans that are released unintentionally, e.g., epizootic vaccinia and transmissible vaccine-derived polioviruses |
Variables in Disease Emergence: The Agent, Host, and Environment

The Role of the Infectious Agent in the Emergence of Infectious Diseases

The Role of the Host in the Emergence of Infectious Diseases
The role of the environment in the emergence of infectious diseases, emergence of diseases leading to epidemicity and endemicity.
| Respiratory including Environmental | Gastrointestinal including Environmental | Inoculation | |
|---|---|---|---|
| Direct | Vectorborne | ||
| Influenza | Cholera | Anthrax | Chikungunya |
| Human coronaviruses | Noroviruses | Dracunculiasis | Dengue |
| Measles | Rotaviruses | Gonorrhea | Lyme disease |
| Rhinoviruses | Salmonellosis | Hepatitis B and C | Malaria |
| SARS, COVID-19, MERS | Typhoid fever | HIV | Yellow fever |
| Some human enteroviruses | Some human enteroviruses | Syphilis | Zika |
The Enigma of Host-Switching

Summary and Conclusions
Acknowledgments, article metrics, related articles.
- Download Hi-res image
- Download .PPT
- Cancer Cell
- Cell Chemical Biology
- Cell Genomics
- Cell Host & Microbe
- Cell Metabolism
- Cell Reports
- Cell Reports Medicine
- Cell Stem Cell
- Cell Systems
- Current Biology
- Developmental Cell
- Molecular Cell
- American Journal of Human Genetics ( partner )
- Biophysical Journal ( partner )
- Biophysical Reports ( partner )
- Human Genetics and Genomics Advances ( partner )
- Molecular Plant ( partner )
- Molecular Therapy ( partner )
- Molecular Therapy Methods & Clinical Development ( partner )
- Molecular Therapy Nucleic Acids ( partner )
- Molecular Therapy Oncology ( partner )
- Plant Communications ( partner )
- Stem Cell Reports ( partner )
- Trends in Biochemical Sciences
- Trends in Cancer
- Trends in Cell Biology
- Trends in Ecology & Evolution
- Trends in Endocrinology & Metabolism
- Trends in Genetics
- Trends in Immunology
- Trends in Microbiology
- Trends in Molecular Medicine
- Trends in Neurosciences
- Trends in Parasitology
- Trends in Pharmacological Sciences
- Trends in Plant Science
- Cell Reports Physical Science
- Chem Catalysis
- Trends in Chemistry
- Cell Biomaterials
- Cell Reports Methods
- Cell Reports Sustainability
- STAR Protocols
- Nexus ( partner )
- The Innovation ( partner )
- Trends in Biotechnology
- Trends in Cognitive Sciences
- Submit article
- Multi-Journal Submission
- STAR Methods
- Sneak Peek – Preprints
- Information for reviewers
- Cell Symposia
- Consortia Hub
- Cell Press Podcast
- Cell Press Videos
- Coloring and Comics
- Cell Picture Show
- Research Arc
- About Cell Press
- Open access
- Sustainability hub
- Inclusion and diversity
- Help & Support
- Cell Press Careers
- Scientific job board
- Read-It-Now
- Recommend to Librarian
- Publication Alerts
- Best of Cell Press
- Cell Press Reviews
- Cell Press Selections
- Nucleus Collections
- SnapShot Archive
- For Advertisers
- For Recruiters
- For Librarians
- Privacy Policy
- Terms and Conditions
- Accessibility
The content on this site is intended for healthcare professionals and researchers across all fields of science.
We use cookies to help provide and enhance our service and tailor content. To update your cookie settings, please visit the Cookie settings for this site. All content on this site: Copyright © 2024 Elsevier Inc., its licensors, and contributors. All rights are reserved, including those for text and data mining, AI training, and similar technologies. For all open access content, the Creative Commons licensing terms apply.
- Privacy Policy
- Terms & Conditions
- Accessibility
- Help & Support

Session Timeout (2:00)
Your session will expire shortly. If you are still working, click the ‘Keep Me Logged In’ button below. If you do not respond within the next minute, you will be automatically logged out.
- Search Close search
- Find a journal
- Search calls for papers
- Journal Suggester
- Open access publishing
We’re here to help
Find guidance on Author Services
Open access
Epidemiology and global spread of emerging tick-borne Alongshan virus
- Cite this article
- https://doi.org/10.1080/22221751.2024.2404271
Introduction
Alongshan virus circulation, genetic diversity of alongshan virus, alongshan virus and human health, influences of global warming and urbanization on viral spread, concluding remarks.
- Supplemental material
- Acknowledgements
- Full Article
- Figures & data
- Supplemental
- Reprints & Permissions
- View PDF PDF View EPUB EPUB
The emergence and spread of novel viral pathogens is a major threat to human health, particularly in the context of climate and human-induced change in land use. Alongshan virus (ALSV) is a tick-borne virus associated with human disease, which was first identified in northeast China. More recently, several studies reported the emergence of ALSV in mammalian and arthropod hosts in multiple different countries outside of Asia, and the first viral genome sequencing data has become available. ALSV is a member of the Jingmenvirus group closely related to the Flaviviridae family. Unusually, the positive-sense, single-stranded RNA genome of ASLV is segmented and consists of four distinct segments, two of which show homology with the NS3 and NS5 protein encoding regions of non-segmented flaviviruses. Transmission of arthropod-borne pathogens will likely increase in the future due to environmental change mediated by a variety of environmental and ecological factors and increasing human encroachment into wild animal habitats. In this review, we present current knowledge of global ALSV distribution and emergence patterns, highlight genetic diversity, evolution and susceptible species. Finally, we discuss the role of this emerging tick-borne virus in the context of urbanization and global health.
- Alongshan virus
- segmented flavi-like viruses
- epidemiology
- urbanization
- climate change
According to the World Health Organization, vector-borne diseases make up more than 17% of infectious diseases worldwide and account for more than 700.000 deaths per year (Source: WHO, see https://www.who.int/news-room/fact-sheets/detail/vector-borne-diseases ). As these pathogens and their respective vectors can spread dynamically and rapidly, they constitute a considerable burden and threat to global health. In addition, the spread of vector-borne diseases is fueled by habitat changes caused by climate change or anthropogenic factors and by human encroachment into wildlife habitats. Amongst the pathogens with increased risk for causing human pandemics are Flaviviruses.
Flaviviridae are a family of small enveloped viruses with positive-sense RNA genomes of approximately 9.0–13 kb [1]. Known for their high genomic and phenotypic variability, members of the Flaviviridae family infect a wide range of mammals, birds, and arthropods and thus have a high socio-economic impact. This family can be divided into four genera with distinct characteristics: Hepacivirus, Pegivirus, Pestivirus and Orthoflavivirus. The majority of members of the genus Orthoflavivirus are arthropod-borne and represent important human and veterinary pathogens ( e.g., yellow fever virus, dengue virus, West Nile virus) [2,3]. Despite the substantial sequence divergence between the genera within the Flaviviridae , these viruses exhibit a similar genomic structure characterized by a single open reading frame (ORF) flanked by 5′- and 3′-terminal non-coding regions (NCRs). Structural and nonstructural viral proteins are synthesized as part of a polyprotein that is co- and post-translationally cleaved by viral and cellular proteases. Among the nonstructural protein products, NS3 and NS5 encode the enzymatic domains essential for RNA capping and genome replication, whereas the NS3 and NS2b proteins form a two-component serine protease involved in posttranslational cleavage of the viral polyprotein [4,5]. In 2014, the textbook knowledge of the Flaviviridae genomic structure had been revised, when Qin et al. reported the discovery of the first segmented flavi-like virus in ticks, which was subsequently named Jingmen tick virus (JMTV) [6]. JMTV was shown to have a plus-stranded RNA genome composed of four distinct fragments: S1, S2, S3, and S4. Fragments S1 and S3 encode nonstructural proteins (NSP1 and NSP2), which are homologous to NS3 and NS5 of other flaviviruses. The fragments S2 and S4 encode structural proteins (VP1, VP2, and VP3), which present no homology with any known viral sequence outside the Jingmenvirus group to date [6,7]. JMTV has been detected in several tick species as well as mosquitos, rodents, cattle, goats, bats and primates [8]. More recently, JMTV genome sequences were detected in human fatal cases of Crimean-Congo haemorrhagic fevervirus infection in Kosovo [9] and in four individuals from China [10]. These patients presented with an itchy or painful eschar at the site of tick bite including fever, headache, and myalgia, demonstrating that JMTV can infect humans and manifest clinically. As a result of tick-borne disease surveillance in several countries, another unknown virus, termed Alongshan virus (ALSV), has recently been identified in a patient with unexplained febrile illness in Inner Mongolia in 2017 [11]. This virus demonstrated a high sequence identity to JMTV and has since then been detected in multiple mammals and arthropods and in additional countries across the globe. In this review, we will provide an overview of the current knowledge on ALSV, including its circulation and genetic diversity. Furthermore, we will discuss zoonotic features of ALSV and its impact on global health in the context of urbanization and global warming.
Figure 1. (A) Alongshan virus (ALSV) epidemiology. 272 sequences were downloaded from NCBI, including metadata from sampling locations. In conjunction with reports of ALSV prevalence across Asia and Europe, we summarise in which countries the virus has been detected. (B) Flavivirus particle structure and ALSV and Jingmentick virus genome organisation. The genomes are divided into four segments (S1 - S4): S1 encodes the NS5-like protein, segment 2 encodes the glycoproteins - monocistronic for JMTV and bicistronic for ALSV. S3 encodes an NS3-like protein and S4 encodes the capsid and the VP3 protein. Icons have been retrieved from phylopic.org.

Table 1: Global distribution of ALSV, its host range and testing methods for genome or antibody detection. IFA, immunofluorescence assay.
In summary, ALSV has been detected in a wide range of hosts, which is consistent with the host-range of ticks from the Ixodidae family that are thought to be the main reservoir of ALSV. However, with limited studies investigating ALSV distribution using different methodologies, the overall prevalence remains difficult to estimate. It is recommended that future ALSV prevalence assessments be complemented by a standardized methodology that includes reference sequences and commercial kits for the detection and quantification of nucleic acids and antibodies. In addition, these could be complemented by pan-Jingmenvirus detection methods, which could provide additional insight into the diversity of these viruses. In addition, it may be relevant to examine sentinel animals that have regular contact with ticks, which can provide valuable information on the presence of ALSV.
Despite the fact that flaviviruses display differences in host reservoirs, species tropism, and genetic composition, members of this family have been traditionally believed to have a similar genome structure. This positive sense ssRNA genome encodes a single open reading frame (ORF) and lacks a polyadenylation signal. The encoded ORF is translated as single polyprotein which is post-translationally cleaved into structural and non-structural (NS) proteins. After the discovery of JMTV in 2014, this paradigm has become obsolete. The ALSV genome, like the majority of viruses in the Jingmenvirus group, is organized in four different polyadenylated segments (S1 - S4, see Figure 1 B, Box1) with similar untranslated regions [6]. Although most segmented flavi-like viruses, including ALSV, share this genomic structure, others – e.g. Guaico Culex virus – possess 5 genome segments and lack polyadenylation [26]. Despite these differences in genome architecture, ALSV segment 1 and 3 encoded proteins share high degrees of homology with NS5 and NS3 of prototypic flaviviruses, respectively [11]. Segment 1 encodes a NS5-like protein sharing 26% identical (and 52% chemically similar) amino acids with a 814 amino acid stretch of the yellow fever virus (YFV) NS5 RNA-dependent RNA polymerase (RdRp) ( Figure 1 B) [7]. Despite relatively low sequence similarity, structural and functional homology of the NS5 domain between ALSV and canonical flaviviruses has been demonstrated. This includes its three-dimensional protein folding and the substrate binding properties of the methyltransferase domain [27]. Segment 3 encodes a NS3-like protein with 23% amino acid homology (and 52% chemical amino acid similarity) to a 508 amino acid segment within YFV NS3 serine protease and helicase domains [7]. Similarly, despite the low level of sequence homology, the overall structure, including the ATPase active site and the RNA binding groove, appears to be conserved between ALSV and canonical flaviviruses [28]. Since the enzymatic function of the methyltransferase, RdRp, protease or helicase is essential for flaviviral replication, these proteins may facilitate the possibility of antiviral drug design or drug repurposing. Segment 2 and 4 encoded proteins show fewer similarities with the counterparts of prototypical flaviviruses and appear to be more distantly related to them. They also appear to diverge within the Jingmenvirus group, particularly segment 2, which encodes envelope glycoproteins of Jingmenviruses. In JMTV, these glycoproteins are encoded by a single ORF (VP1), whereas in ALSV they are bicistronic, encoding the glycoproteins VP1a and VP1b ( Figure 1 B). As these glycoproteins initially showed no apparent homology to typical flaviviruses envelope proteins or those of other viruses, as determined by BLASTx or BLASTp, it was initially assumed that they were unrelated. More recently, however, Garry and Garry et al. have shown that a small region of 190 amino acids within these glycoproteins resembles other flaviviruses, containing 23% identical and 50% chemically similar amino acids [7]. In addition, they suggested that the glycoproteins of viruses from the Jingmenvirus group have diverged from a common class II viral fusion protein ancestor. As these divergent evolutionary signatures exist both between prototype flaviviruses and the Jingmenvirus group, as well as within the Jingmenvirus group, new questions arise about their evolutionary history, cell tropism, and mode of infection. The fourth segment (S4) encodes the viral capsid protein, which has no close homology to any proteins of other flaviviruses. This segment also encodes viral protein 3 (VP3), a predicted membrane protein who’s function is currently unknown. Between JMTV and ALSV, S4 is relatively well conserved with ∼75% similarity at the amino acid level. Although some proteins exhibit high conservation between ALSV and other members of the Jingmenvirus group as well as canonical flaviviruses, our understanding of the cellular processes engaged in ALSV replication or pathology remains limited. The ectopic expression of ALSV proteins demonstrated similarities in the cellular localization of some viral proteins to the endoplasmic reticulum (ER), a key feature of many flaviviruses [29]. Moreover, ectopic expression of NSP1 was observed to diminish the quantity of mitochondria, indicating potential mechanisms for virus-induced pathology. Nevertheless, further investigation is essential to gain a deeper comprehension of the molecular mechanisms underlying Jingmen virus replication and pathology, with the aim of utilizing them for the development of novel antiviral therapeutics.
Figure 2. Phylogenetic analysis of ALSV genome segments. (A) segment 1, (B) segment 2, (C) segment 3 and (D) segment 4. Phylogenetic analysis was conducted using publicly available data downloaded from NCBI (n = 272, Table S1). For each segment, Shannon entropy and alignment coverage was calculated (top panel). Partial nucleotide sequences (n = 144) from the highlighted area (dotted line) were aligned using clustal omega and subsequently used to construct phylogenetic trees. Trees (below) were generated using the maximum likelihood method implemented in IQtree2 with the best model finder option. Values assigned to deep internal nodes (green circles) within the phylogeny represent bootstrap supports above 80 from 1,000 repetitions. Each tree was normalized to the scale of 0.05 mutations per site. Branch color highlighting indicates sample origin. Icons denoted at the tips indicate host species. Deep divergences within the tree resulted in three generally well supported clusters denoted as C1, C2, C3. Within these, C1 derived sequences originated from Europe, C2 sequences from middle Asia and C3 from eastern parts of Asia. Icons have been retrieved from phylopic.org.

To date, the incidence and prevalence of tick-borne ALSV infection in humans remains difficult to estimate, as data on human infections have only been collected in China [11]. However, the fact that ALSV has been detected in tick populations in close proximity to humans is a good example of the consequences of urbanization and raises concerns about future outbreaks. Indeed, a vast majority of the 86 patients that acquired ALSV infection in China had been working outdoors in fields, forests, or agriculture prior to the tick bite and the incubation period until onset of disease spanned from one to 10 days. Infection with ALSV in these patients caused febrile illness with unspecific symptoms, resembling infections caused by other vector-transmitted Jingmenviruses [11]. The most common clinical symptoms were fever, headaches, and fatigue, followed by other non-specific symptoms such as depression, coma, nausea, myalgia/arthralgia, and rashes. With limited knowledge on ALSV infection, however, clinical diagnosis alone can prove to be challenging and requires a combination of more extensive testing methods. These include measurements of routine laboratory parameters such as lactate dehydrogenase (LDH), C-reactive protein (CRP), creatine kinase (CK), and liver transaminases, as well as measurements of cerebrospinal fluid (CSF) parameters. In infected patients, elevated levels of C-reactive protein and lactate dehydrogenase were observed in 50% and 68%, respectively, while elevated liver transaminases were observed in 25-29% of cases and creatine kinase levels were elevated in 9% of cases [11]. Radiological imaging revealed ischemic white matter demyelination in the brain in 7 out of 54 (13%) cases and one patient showed elevated protein and white-cell count in the CSF. Additionally, serological analyses in the active and convalescent phases of infection have also been performed to detect human ALSV infection using immunofluorescence and microneutralization assays, where seroconversion was defined as at least fourfold increased levels of virus-specific IgG in the convalescent phase. Furthermore, nucleic acid amplification (RT-PCR) from whole-blood samples using virus-specific primers has been used to detect ALSV virus infection in human patients. Although the circulation of ALSV at the human tick interface is still poorly understood, methods to detect this emerging pathogen should be improved to control future outbreaks.
None of the symptomatic ALSV infections resulted in long-term damage or death. The clinical symptoms resolved within 6 to 8 days under empirical treatment with ribavirin and benzylpenicillin and the duration of hospitalization ranged from 10 to 14 days [11]. So far, no proof of transmission between ALSV-infected and non-infected humans exists. However, vector-borne flaviviruses have in the past caused epidemics with significant public health impact [32–34]. Therefore, the development and implementation of sufficiently effective preventative measures such as vector monitoring, diagnostic tools, and therapeutic strategies, are essential to contain potential future outbreaks/epidemics. This includes surveillance of circulating strains, research on the molecular properties of emerging virus groups, the administration of vaccines or antiviral drugs, as well as other disease-specific measures, which have been undertaken to help combat the vector-borne flavivirus disease burden [35–37]. Moreover, medical information, public education and awareness play a significant part in the spread and control of vector-borne disease.
The clinical presentation of ALSV infection is reminiscent of other tick-borne viruses causing nonspecific febrile disease. While a clear tissue tropism of ALSV infection has not yet been established, the documented systemic symptoms including fever, headache, fatigue, coma, and nausea might imply a central nervous-tropism. Therefore, it could be speculated that ALSV possesses properties and virulence factors similar to tick-borne flaviviruses which cause encephalitic disease. Tick-borne flaviviruses usually present in one of two general phenotypes: haemorrhagic fever or encephalitic disease. The pathogenicity of TBE viruses in humans, including their neuroinvasiveness and neurovirulence, is mediated by virulence factors including structural proteins, non-structural proteins, and untranslated regions (UTRs) of the viral mRNA, among others [38]. These factors play a role in determining processes such as cell entry, replication, as well as antigenicity and immune evasion. The NS5 protein of TBEV, e.g., has been linked to the development of neurological disease phenotypes of the virus in mice [39]. At present, the extent to which such factors may also be relevant for Alongshan virus is still unclear and requires further investigation. Similar to other flaviviruses, however, ALSV appears to employ strategies to circumvent host defense mechanisms, e.g. by interfering with IFN-β-mediated ISG induction through degradation of STAT2 [40]. Identification of pathogenic mechanisms, including host-virus interactions, will be crucial for understanding and combating ALSV infections in humans.
Figure 3. Tick-borne virus-human interface. Climate change and urbanization may increase the vector-human interface in the future. Potential interfaces include urban green spaces, parks, suburban areas and outdoor activities. Ticks can amplify in these and potentially transmit diseases.

Particularly, across an urban-to-rural gradient, landscape configuration and habitat connectivity, e.g. of urban green spaces, have been shown to directly impact tick abundance and tick-borne disease incidence through alteration of the population, behavior, or physiology of ticks and their host species [62–64]. For example, Ixodes tick parasitism has been demonstrated to be reduced in urban bird populations in eastern France [65]. While most abundant in rural areas and significantly reduced numbers in sampling sites around urbanized areas in Sweden, Ixodes ricinus presence could also be recorded in green spaces within highly urbanized areas, albeit to a lesser degree [66]. Host species that are able to adapt to the increasingly fragmented, urbanized environments represent important players in tick maintenance and pathogen reservoirs and increase transmission risk of tick-borne disease to humans in these otherwise less-favorable environments [67].
In addition to bacterial pathogens, Flaviviridae appear among the most prominent infectious agents transmitted by the two key vector types, mosquitoes and ticks [61]. The newly emerging, segmented flavi-like viruses have attracted particular interest lately. The epidemiology and risk of infection of ALSV are likely to be affected by the same mechanisms, similar to other tick-borne viruses such as TBEV [51,68]. Understanding these complex dynamics of anthropogenic land use changes and the influence on the tick-host-predator system is, therefore, crucial for risk assessment and reduction of emerging tick bite-associated diseases, including ALSV and similar vector-borne viruses.
Due to climate change, globalization and urbanization, emerging viruses present a constant threat to global health. In particular, spill overs of viruses that are transmitted through arthropod vectors are becoming more frequent as the wildlife human interface increases, highlighting the significance to human health. The emergence of evolutionary distinct viruses, such as ALSV, presents a new and understudied risk. Therefore, it is crucial to examine the genomic diversity and prevalence of these viruses to prepare for potential spill overs that may occur in the near future and establish preventive measures.
Funding statement:
E.S was supported by a Grant of the German Centre for Infection Research (DZIF).
Authors’ contributions:
A.G., A.L., E.S. designed the review outline and conducted primary literature search. A.G., S.J., E.S. illustrated the figures. J.S. and E.S. supervised. All authors contributed to drafting the text and approved the manuscript. They agree, that they are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Box 1. Alongshan virus at a glance.
Phylogenetic Flavi-like virus
classification: Related to Orthoflavivirus, but
remains as unclassified
Jingmenvirus group
Genome: ssRNA, 4 Segments (S1 - S4)
S1: NS5-like RdRp, 2988 bp
S2: Glycoproteins, 2752 bp
S3: NS3-like protease, 2721 bp
S4: Core and VP3, 2701 bp
Host range: Arthropods and mammals
Virion: Spherical, 80–100 nm, enveloped
Symptoms: Fever, headaches, fatigue
Treatment: no specific treatment ribavirin (off-label)
20240724_ALSVreviewSupplements.docx
Acknowledgements :.
We thank the whole team at the Department of Molecular and Medical Virology and especially Mara Klöhn, Richard Brown and Stephanie Pfänder for careful reading of the paper and their insightful comments.
- Simmonds P, Becher P, Bukh J, et al. ICTV Virus Taxonomy Profile: Flaviviridae. Journal of General Virology. 2017. p. 2–3. Google Scholar
- Pierson TC, Diamond MS. The continued threat of emerging flaviviruses. Nat Microbiol. 2020;5:796–812. Google Scholar
- Genus: Orthoflavivirus | ICTV [Internet]. [cited 2024 Jan 20]. Available from: https://ictv.global/report/chapter/flaviviridae/flaviviridae/orthoflavivirus . Google Scholar
- Bollati M, Alvarez K, Assenberg R, et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antiviral Research. 2010;87:125–148. Google Scholar
- Davidson AD. Chapter 2 New Insights into Flavivirus Nonstructural Protein 5. Advances in Virus Research [Internet]. Academic Press; 2009 [cited 2024 Jan 31]. p. 41–101. Available from: https://www.sciencedirect.com/science/article/pii/S0065352709740023 . Google Scholar
- Qin X-C, Shi M, Tian J-H, et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proceedings of the National Academy of Sciences. 2014;111:6744–6749. Google Scholar
- Garry CE, Garry RF. Proteomics Computational Analyses Suggest That the Envelope Glycoproteins of Segmented Jingmen Flavi-Like Viruses are Class II Viral Fusion Proteins (β-Penetrenes) with Mucin-Like Domains. Viruses. 2020;12:260. Google Scholar
- Colmant AMG, Charrel RN, Coutard B. Jingmenviruses: Ubiquitous, understudied, segmented flavi-like viruses. Front Microbiol. 2022;13:997058. Google Scholar
- Emmerich P, Jakupi X, von Possel R, et al. Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever orthonairovirus in humans, Kosovo. Infection, Genetics and Evolution. 2018;65:6–11. Google Scholar
- Jia N, Liu H-B, Ni X-B, et al. Emergence of human infection with Jingmen tick virus in China: A retrospective study. EBioMedicine. 2019;43:317–324. Google Scholar
- Wang Z-D, Wang B, Wei F, et al. A New Segmented Virus Associated with Human Febrile Illness in China. N Engl J Med. 2019;380:2116–2125. Google Scholar
- Wu Z, Zhang M, Zhang Y, et al. Jingmen tick virus: an emerging arbovirus with a global threat. mSphere. 2023;8:e00281-23. Google Scholar
- Wang Z-D, Wang W, Wang N-N, et al. Prevalence of the emerging novel Alongshan virus infection in sheep and cattle in Inner Mongolia, northeastern China. Parasites & Vectors. 2019;12:450. Google Scholar
- Kuivanen S, Levanov L, Kareinen L, et al. Detection of novel tick-borne pathogen, Alongshan virus, in Ixodes ricinus ticks, south-eastern Finland, 2019. Eurosurveillance. 2019;24:1900394. Google Scholar
- Kholodilov IS, Litov AG, Klimentov AS, et al. Isolation and Characterisation of Alongshan Virus in Russia. Viruses. 2020;12:362. Google Scholar
- Kholodilov IS, Belova OA, Morozkin ES, et al. Geographical and Tick-Dependent Distribution of Flavi-Like Alongshan and Yanggou Tick Viruses in Russia. Viruses. 2021;13:458. Google Scholar
- Kholodilov IS, Belova OA, Ivannikova AY, et al. Distribution and Characterisation of Tick-Borne Flavi-, Flavi-like, and Phenuiviruses in the Chelyabinsk Region of Russia. Viruses. 2022;14:2699. Google Scholar
- Stanojević M, Li K, Stamenković G, et al. Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia. Viruses. 2020;12:975. Google Scholar
- Ebert CL, Söder L, Kubinski M, et al. Detection and Characterization of Alongshan Virus in Ticks and Tick Saliva from Lower Saxony, Germany with Serological Evidence for Viral Transmission to Game and Domestic Animals. Microorganisms. 2023;11:543. Google Scholar
- Stegmüller S, Fraefel C, Kubacki J. Genome Sequence of Alongshan Virus from Ixodes ricinus Ticks Collected in Switzerland. Microbiology Resource Announcements. 2023;12:e01287-22. Google Scholar
- Stegmüller S, Qi W, Torgerson PR, et al. Hazard potential of Swiss Ixodes ricinus ticks: Virome composition and presence of selected bacterial and protozoan pathogens. PLOS ONE. 2023;18:e0290942. Google Scholar
- Temmam S, Bigot T, Chrétien D, et al. Insights into the Host Range, Genetic Diversity, and Geographical Distribution of Jingmenviruses. mSphere. 2019;4:e00645-19. Google Scholar
- Makino Y, Tadano M, Saito M, et al. Studies on Serological Cross-Reaction in Sequential Flavivirus Infections. Microbiology and Immunology. 1994;38:951–955. Google Scholar
- Lalita Priyamvada RA William Hudson, Wrammert J. Humoral cross-reactivity between Zika and dengue viruses: implications for protection and pathology. Emerging Microbes & Infections. 2017;6:1–6. Google Scholar
- Chan KR, Ismail AA, Thergarajan G, et al. Serological cross-reactivity among common flaviviruses. Frontiers in Cellular and Infection Microbiology [Internet]. 2022 [cited 2024 Feb 18];12. Available from: https://www.frontiersin.org/articles/10.3389fcimb.2022.975398 . Google Scholar
- Ladner JT, Wiley MR, Beitzel B, et al. A Multicomponent Animal Virus Isolated from Mosquitoes. Cell Host Microbe. 2016;20:357–367. Google Scholar
- Chen H, Lin S, Yang F, et al. Structural and functional basis of low-affinity SAM/SAH-binding in the conserved MTase of the multi-segmented Alongshan virus distantly related to canonical unsegmented flaviviruses. PLOS Pathogens. 2023;19:e1011694. Google Scholar
- Gao X, Zhu K, Wojdyla JA, et al. Crystal structure of the NS3-like helicase from Alongshan virus. IUCrJ. 2020;7:375–382. Google Scholar
- Zhao Y, Wu P, Liu L, et al. Characterization and subcellular localization of Alongshan virus proteins. Frontiers in Microbiology [Internet]. 2022 [cited 2023 Nov 25];13. Available from: https://www.frontiersin.org/articles/10.3389fmicb.2022.1000322 . Google Scholar
- Guo J-J, Lin X-D, Chen Y-M, et al. Diversity and circulation of Jingmen tick virus in ticks and mammals. Virus Evol. 2020;6:veaa051. Google Scholar
- Ecker M, Allison SL, Meixner T, et al. Sequence analysis and genetic classification of tick-borne encephalitis viruses from Europe and Asia. Journal of General Virology. 1999;80:179–185. Google Scholar
- Carteaux G, Maquart M, Bedet A, et al. Zika Virus Associated with Meningoencephalitis. N Engl J Med. 2016;374:1595–1596. Google Scholar
- Halstead SB. Reappearance of chikungunya, formerly called dengue, in the Americas. Emerg Infect Dis. 2015;21:557–561. Google Scholar
- Kyle JL, Harris E. Global spread and persistence of dengue. Annu Rev Microbiol. 2008;62:71–92. Google Scholar
- Shaw WR, Catteruccia F. Vector biology meets disease control: using basic research to fight vector-borne diseases. Nat Microbiol. 2019;4:20–34. Google Scholar
- Dutra HLC, Rocha MN, Dias FBS, et al. Wolbachia Blocks Currently Circulating Zika Virus Isolates in Brazilian Aedes aegypti Mosquitoes. Cell Host Microbe. 2016;19:771–774. Google Scholar
- Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell. 2009;139:1268–1278. Google Scholar
- Kellman EM, Offerdahl DK, Melik W, et al. Viral Determinants of Virulence in Tick-Borne Flaviviruses. Viruses. 2018;10:329. Google Scholar
- Yoshii K, Sunden Y, Yokozawa K, et al. A Critical Determinant of Neurological Disease Associated with Highly Pathogenic Tick-Borne Flavivirus in Mice. Journal of Virology. 2014;88:5406–5420. Google Scholar
- Zhao Y, Sui L, Pan M, et al. The segmented flavivirus Alongshan virus reduces mitochondrial mass via degrading STAT2 to suppress innate immune response. bioRxiv [Internet]. 2024; Available from: https://www.biorxiv.org/content/early/2024/03/07/2024.03.06.583679 . Google Scholar
- Reisen WK. Landscape epidemiology of vector-borne diseases. Annu Rev Entomol. 2010;55:461–483. Google Scholar
- Reisen WK, Fang Y, Martinez VM. Effects of temperature on the transmission of west nile virus by Culex tarsalis (Diptera: Culicidae). J Med Entomol. 2006;43:309–317. Google Scholar
- Pfäffle M, Littwin N, Muders SV, et al. The ecology of tick-borne diseases. International Journal for Parasitology. 2013;43:1059–1077. Google Scholar
- Ni X-B, Pei Y, Ye Y-T, et al. Eco-climate drivers shape virome diversity in a globally invasive tick species. ISME J. 2024;wrae087. Google Scholar
- Bernard C, Kukla CJ, Rakotoarivony I, et al. Detection of Crimean–Congo haemorrhagic fever virus in Hyalomma marginatum ticks, southern France, May 2022 and April 2023. Eurosurveillance. 2024;29:2400023. Google Scholar
- Lindgren E, Tälleklint L, Polfeldt T. Impact of climatic change on the northern latitude limit and population density of the disease-transmitting European tick Ixodes ricinus. Environ Health Perspect. 2000;108:119–123. Google Scholar
- Jore S, Viljugrein H, Hofshagen M, et al. Multi-source analysis reveals latitudinal and altitudinal shifts in range of Ixodes ricinus at its northern distribution limit. Parasites & Vectors. 2011;4:84. Google Scholar
- Daniel M, Danielová V, Kříž B, et al. Shift of the Tick Ixodes ricinus and Tick-Borne Encephalitis to Higher Altitudes in Central Europe. Eur J Clin Microbiol Infect Dis. 2003;22:327–328. Google Scholar
- Jaenson TG, Hjertqvist M, Bergström T, et al. Why is tick-borne encephalitis increasing? A review of the key factors causing the increasing incidence of human TBE in Sweden. Parasites & Vectors. 2012;5:184. Google Scholar
- Tokarevich NK, Tronin AA, Blinova OV, et al. The impact of climate change on the expansion of Ixodes persulcatus habitat and the incidence of tick-borne encephalitis in the north of European Russia. Global Health Action. 2011;4:8448. Google Scholar
- Korenberg E, Cerný V, Daniel M. Occurrence of ixodid ticks--the main vectors of tick-borne encephalitis virus in urbanized territory. Folia Parasitol (Praha). 1984;31:365–370. Google Scholar
- Anis H, Basha Shaik A, Karabulut E, et al. Upsurge of Powassan virus disease in northeastern United States: a public health concern—a short communication. Ann Med Surg (Lond). 2023;85:5823–5826. Google Scholar
- Gray JS, Dautel H, Estrada-Peña A, et al. Effects of climate change on ticks and tick-borne diseases in europe. Interdiscip Perspect Infect Dis. 2009;2009:593232. Google Scholar
- Mahon MB, Sack A, Aleuy OA, et al. A meta-analysis on global change drivers and the risk of infectious disease. Nature. 2024;629:830–836. Google Scholar
- Shochat E, Lerman SB, Anderies JM, et al. Invasion, Competition, and Biodiversity Loss in Urban Ecosystems. BioScience. 2010;60:199–208. Google Scholar
- McKinney ML. Urbanization, Biodiversity, and Conservation: The impacts of urbanization on native species are poorly studied, but educating a highly urbanized human population about these impacts can greatly improve species conservation in all ecosystems. BioScience. 2002;52:883–890. Google Scholar
- Mackenzie JS, Gubler DJ, Petersen LR. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med. 2004;10:S98–S109. Google Scholar
- Brown H, Duik-Wasser M, Andreadis T, et al. Remotely-sensed vegetation indices identify mosquito clusters of West Nile virus vectors in an urban landscape in the northeastern United States. Vector Borne Zoonotic Dis. 2008;8:197–206. Google Scholar
- Diuk-Wasser MA, VanAcker MC, Fernandez MP. Impact of Land Use Changes and Habitat Fragmentation on the Eco-epidemiology of Tick-Borne Diseases. J Med Entomol. 2021;58:1546–1564. Google Scholar
- Stoddard ST, Morrison AC, Vazquez-Prokopec GM, et al. The Role of Human Movement in the Transmission of Vector-Borne Pathogens. PLOS Neglected Tropical Diseases. 2009;3:e481. Google Scholar
- Swei A, Couper LI, Coffey LL, et al. Patterns, Drivers, and Challenges of Vector-Borne Disease Emergence. Vector-Borne and Zoonotic Diseases. 2020;20:159–170. Google Scholar
- VanAcker MC, Little EAH, Molaei G, et al. Enhancement of Risk for Lyme Disease by Landscape Connectivity, New York, New York, USA. Emerg Infect Dis. 2019;25:1136–1143. Google Scholar
- Jackson LE, Hilborn ED, Thomas JC. Towards landscape design guidelines for reducing Lyme disease risk. Int J Epidemiol. 2006;35:315–322. Google Scholar
- Estrada-Peña A. Understanding the Relationships between Landscape Connectivity and Abundance of Ixodes ricinus Ticks. Exp Appl Acarol. 2002;28:239–248. Google Scholar
- Gregoire A, Faivre B, Heeb P, et al. A comparison of infestation patterns by Ixodes ticks in urban and rural populations of the Common Blackbird Turdus merula. Ibis. 2002;144:640–645. Google Scholar
- Janzén T, Hammer M, Petersson M, et al. Factors responsible for Ixodes ricinus presence and abundance across a natural-urban gradient. PLOS ONE. 2023;18:e0285841. Google Scholar
- Rizzoli A, Silaghi C, Obiegala A, et al. Ixodes ricinus and Its Transmitted Pathogens in Urban and Peri-Urban Areas in Europe: New Hazards and Relevance for Public Health. Front Public Health. 2014;2:251. Google Scholar
- Danielová V, Daniel M, Schwarzová L, et al. Integration of a tick-borne encephalitis virus and Borrelia burgdorferi sensu lato into mountain ecosystems, following a shift in the altitudinal limit of distribution of their vector, Ixodes ricinus (Krkonose mountains, Czech Republic). Vector Borne Zoonotic Dis. 2010;10:223–230. Google Scholar
- Xu W, Wang W, Li L, et al. Alongshan Virus Infection in Rangifer tarandus Reindeer, Northeastern China - Volume 30, Number 7—July 2024 - Emerging Infectious Diseases journal - CDC. [cited 2024 Jul 1]; Available from: https://wwwnc.cdc.gov/eid/article/30/7/23-1219_article . Google Scholar
- Back to Top
Related research
People also read lists articles that other readers of this article have read.
Recommended articles lists articles that we recommend and is powered by our AI driven recommendation engine.
Cited by lists all citing articles based on Crossref citations. Articles with the Crossref icon will open in a new tab.
- People also read
- Recommended articles
To cite this article:
Download citation, your download is now in progress and you may close this window.
- Choose new content alerts to be informed about new research of interest to you
- Easy remote access to your institution's subscriptions on any device, from any location
- Save your searches and schedule alerts to send you new results
- Export your search results into a .csv file to support your research
Login or register to access this feature
Register now or learn more
- Scoping Review
- Open access
- Published: 16 May 2023
Climate change and infectious disease: a review of evidence and research trends
- Paige Van de Vuurst 1 , 2 , 3 &
- Luis E. Escobar ORCID: orcid.org/0000-0001-5735-2750 2 , 3 , 4 , 5
Infectious Diseases of Poverty volume 12 , Article number: 51 ( 2023 ) Cite this article
18k Accesses
21 Citations
36 Altmetric
Metrics details
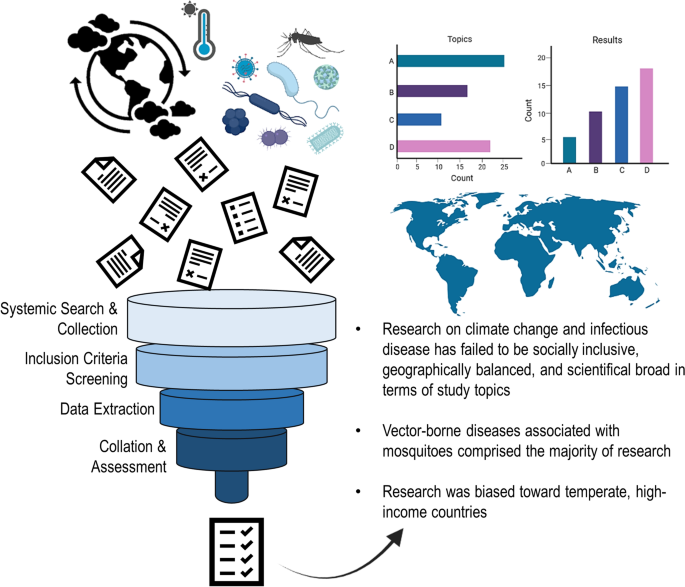
Climate change presents an imminent threat to almost all biological systems across the globe. In recent years there have been a series of studies showing how changes in climate can impact infectious disease transmission. Many of these publications focus on simulations based on in silico data, shadowing empirical research based on field and laboratory data. A synthesis work of empirical climate change and infectious disease research is still lacking.
We conducted a systemic review of research from 2015 to 2020 period on climate change and infectious diseases to identify major trends and current gaps of research. Literature was sourced from Web of Science and PubMed literary repositories using a key word search, and was reviewed using a delineated inclusion criteria by a team of reviewers.
Our review revealed that both taxonomic and geographic biases are present in climate and infectious disease research, specifically with regard to types of disease transmission and localities studied. Empirical investigations on vector-borne diseases associated with mosquitoes comprised the majority of research on the climate change and infectious disease literature. Furthermore, demographic trends in the institutions and individuals published revealed research bias towards research conducted across temperate, high-income countries. We also identified key trends in funding sources for most resent literature and a discrepancy in the gender identities of publishing authors which may reflect current systemic inequities in the scientific field.
Conclusions
Future research lines on climate change and infectious diseases should considered diseases of direct transmission (non-vector-borne) and more research effort in the tropics. Inclusion of local research in low- and middle-income countries was generally neglected. Research on climate change and infectious disease has failed to be socially inclusive, geographically balanced, and broad in terms of the disease systems studied, limiting our capacities to better understand the actual effects of climate change on health.
Graphical abstract
The Intergovernmental Panel on Climate Change has anticipated, with high confidence, that climate change will amplify health threats worldwide [ 1 , 2 ], which is supported by the fact that the life cycles of many infectious agents are inextricably linked to climate [ 1 , 3 , 4 , 5 , 6 ]. Multiple studies have shown that variation in temperature, precipitation, and humidity affects the transmission and distribution of infectious diseases [ 7 , 8 , 9 , 10 ]. Nevertheless, the magnitude, direction, and strength of the impact of climate change upon infectious disease transmission remains unclear [ 3 , 5 , 7 ]. To determine what further research is needed to advance a given field in scientific research it is often necessary to synthesize previous work [ 11 ]. This type of retrospective, systematic analysis of literature in a specific topic or field is referred to as a systematic review. Systematic reviews are a popular and effective method commonly utilized to identify trends and gaps in ongoing research [ 12 ]. Results from systematic reviews and scoping studies, which are often used to map the availability of literature on an specific topic [ 13 , 14 ], can be used to guide future research lines, future policy decisions, and can be particularly useful in scientific fields with emerging evidences, such as epidemiology [ 12 , 13 , 15 , 16 ].
Despite their effectiveness, systematic reviews are noticeably lacking in the literary landscape of anthropogenic climate change research, especially with regard to its impacts on infectious diseases. There is, therefore, a need for a systematic synthesis of recent empirical research assessing disease impacts of climate change. Here, we provide a synthesis of scientific literature on climate change and infectious diseases from recent history. The overall objective of this study was to determine the trends of recent empirical research regarding climate change impacts on infectious diseases and to identify geographic, topical, or taxonomic trends of research. We sought to assess the geographic regions where climate change and disease transmission have been under studied, accounting for both study area and first author affiliation to identify geographic and bibliometric signals. In addition, we assessed the taxa of hosts and transmission types of pathogens studied. Finally, we sought to inform future research avenues, policy, and practices via the trends and impacts identified herein.
Search strategy, inclusion criteria, exclusion criteria
Our search strategy included recovering articles from Web of Science (Clarivate™) [ 17 ] and PubMed™ [ 18 ] literary repositories using a key word search. Keywords included "climate change", "global warming", “greenhouse gas*” (*asterisk used to incorporate all forms of the word. i.e., gas, gases, gaseous), “world warming”, “disease”, “infectious”, “pathogen”, “waterborne”, “water borne”, “food borne”, “vector borne”, “parasite”, and “non-vector borne”. Time range restrictions were set from January of 2015 to December of 2020 to incorporate all publications from the most recent, pre-pandemic five-year period of empirical climate change research. This key word search was limited to journal manuscripts, as the purpose of this study was to analyze original peer-reviewed research. Other literature types such as book chapters, review articles, proceedings papers, or conference abstracts were excluded. Articles were then imported into Endnote citation software, where redundant articles were removed.
After collection we conducted an initial screening of both article titles and abstracts. This initial review allowed for the identification of articles which did not fit within the review criteria. Inclusion criteria were: (1) The manuscript was peer-reviewed and published without retraction, (2) the primary goal of the research was centered on assessing climate change and its repercussions, impacts, effects, association, or influences on disease, infection, transmission, infestation, or illness, (3) the research was original and not a review, (4) the research was descriptive, retrospective, and based on real world systems using non-simulated future-climate data (i.e., present-day and past climate only), (5) the manuscript utilized primary data and (6) the pathogen, parasite, vector, or disease of focus impacted either humans, non-human animals, or both. Each article was reviewed by at least two independent reviewers and was confirmed for inclusion or exclusion based on the inclusion criteria. If the independent reviewers were in disagreement on whether or not the article fit the inclusion criteria, the article was reviewed by a third reviewer. Studies which did not fit this inclusion criteria were flagged and maintained in a separate databased. Studies on plant diseases were not within the scope of this study and therefore were excluded.
Evidence extraction and analysis
We then reviewed the remaining publications in full and conducted evidence extraction of each article to conduct our gap analysis of bibliometric, subject, taxonomic, and geographic trends in research and publication. We gathered descriptive metadata from each article to assess when, where, and by whom the articles were published (e.g., year or publication, journal name, title, authors, etc.). To assess authorship demographics, we recorded the lead author and senior author’s names, pronouns, and institutional affiliation for each publication. Authors’ pronouns were recorded based upon the personal distinctions of each individual author, and the pronouns they chose to use (e.g., she/her, he/him, they/them, em/eir, xem/xyr, etc.) on their institutional or research affiliated websites. We implemented this method to be inclusive of all authors’ identities while maintaining personal privacy [ 19 , 20 ]. If the author did not denote their pronouns in any public way, we recorded their pronouns as “unknown”. We also collected descriptive metadata on the study methods and locations or each article including: (1) study location at the country and continent level, (2) disease host, vector, or pathogen studied, (3) transmission method of each disease studied, (4) primary taxa or taxon of interest (i.e., the taxonomic group of the host or infectious organism or organisms being studied), and (5) spatial scale (e.g., local or inferior to country level, regional, country level, or global). To assess the quality of the included literature, we also recorded and synthesized the conclusions of the sampled articles, and reported these findings based upon the author’s interpretation of their results. We also collected descriptive information on the publication funding or support for each article published in the most recent year included in the review (i.e., 2020) to ascertain current funding sources for the most recent climate change and disease publications. We then compared funding sources with current estimates of country gross domestic product (GDP) from the World Bank World Development Indicators Dataset [ 21 ].
To assess the distribution of the categorical topics of the literature we used a Pearson’s chi-squared ( χ 2 ) test. It has been estimated that approximately 60% of known infectious diseases are zoonotic (i.e., originating in non-human animals) [ 16 , 22 ]. We compared this value (60%) with the proportion of literature which assessed zoonotic diseases to identify if the literature followed this expected proportion. We also used the χ 2 test to identify if the proportion of host species categories studied (humans, wildlife, and livestock) were equal. To assess the geographic distribution of publication demographics, the lead authors’ institutional affiliations were recorded for each publication and assigned to their corresponding countries of origin. Demographic data of study locations and author affiliations were summarized and visualized to detect spatial and temporal patterns of these data using ArcGISpro version 2.9.3 and R version 4.1 [ 23 , 24 , 25 ]. We utilized population data from the United Nations Population Division [ 26 ] for the year 2020 to assess the per-capita research effort by country.
Literature demographics
Our initial key word search resulted in 10,461 articles from both PubMed and Web of Science. A total of 621 research articles (5.9%) fit the inclusion criteria for the 2015–2020 period and were retained for evidence extraction and gap analysis. Within these publications, 109 distinct infectious diseases were identified in relation to climate change research. A small portion of publications ( n = 127) assessed multiple diseases within the same study. Authors of the reviewed articles reported that climate change impacted the disease system being assessed in 59% of the articles. Most of the articles (83.9%) which described climate change impacts reported that climate change increased the prevalence, transmission, or suitability for the disease being studied, while 11.5% of studies reported that climate change decreased the prevalence, transmission, or suitability. Only 7.7% of the assessed articles reported no effect of climate change on the disease system being studied. The review revealed that 32.7% of the articles concluded that climate change could “possibly” or “potentially” impact the disease system being assessed (i.e., the authors did not report a definitive pattern).
Research trends
Infectious diseases which originate from cross-species pathogen transmission of animals to humans (i.e., zoonotic diseases) accounted for most of the studies ( n = 288, 46.4%), significantly more than diseases which do not originate from animal to human cross species transmission ( n = 253, 40.7%), ( χ 2 = 9.97, P = 0.002). Infectious diseases which impact humans were well represented within the literature ( n = 406) ( χ 2 = 114.3, P = 0.0001), while infectious diseases affecting livestock were less represented ( n = 152). Only 116 publications assessed diseases affecting wildlife.
The specific conditions most frequently studied from this sample included vector-borne diseases (Fig. 1 ), such as malaria ( n = 58), dengue fever ( n = 37), and Lyme disease ( n = 22) (Fig. 1 ). Vectors most frequently studied were mosquitoes ( n = 174), ticks ( n = 51), and flies ( n = 14) (Fig. 1 ). Frequently studied environmentally transmitted conditions included food and water-borne diseases, such as diarrheal diseases ( n = 18) and chytridiomycosis ( n = 10) (Fig. 1 ). Studies also focused on diseases hosted by arthropods ( n = 189) and humans ( n = 185) (Fig. 1 ). The third most studied host taxonomic group was non-human mammals ( n = 47), followed by amphibians ( n = 19) and birds ( n = 17) (Fig. 1 ). In terms of study scale, research was conducted at the local, regional, or country levels, with less effort for global-level studies (Fig. 2 ).

Trends in climate change and disease research. Number of publications ( x -axis) from 2015–2020 according to A taxa of host species studied, B transmission type of diseases studied, C vector species studied, and D top 20 most studied diseases from over 100 different diseases studied. Multiple: multiple diseases with multiple transmission types studied in a single article

Bibliometric demographics. A Number of publications ( x -axis) from 2015–2020 when delimited by scale of study. N/A: Studies for which a spatial scale was not applicable (e.g., laboratory-based studies) or for which scale was not specified. B Percentile breakdown of lead author affiliations collated into categories based on the institution’s description (i.e., college or university, governmental organizations or research organization). Other: lead author affiliation institutions which do not fit one of these categories including non-governmental organizations, independent researchers, or private companies not otherwise specified
Publication trends
Bibliometric analysis revealed a greater usage of he/him pronouns for both first and senior authors (Fig. 3 ). We recorded no instances of they/them or other non-binary pronouns by first or senior authors from the articles revised. We also found that study areas and affiliation of lead authors most frequently occurred in the United States, China, the United Kingdom, Canada, and Australia (Figs. 4 , 5 ). Research effort accounting for the country’s population size showed that countries such as Norway, Australia, and Canada have a higher comparative research effort than other countries (Fig. 4 ). Most lead author affiliations were linked to higher education institutions (i.e., universities or colleges), with fewer publications originating from governmental organizations or independent research institutions (Fig. 2 ). University affiliations were frequently located in the United States (e.g., the University of California, Colorado State University, University of Florida), and in China (e.g., Shandong University) (Fig. 5 ). Funding for papers published in 2020 was largely sourced from federal or national institutions (53.3% of articles) or a combination of federal and academic institutions (26.7% of articles), with most of this funding originating in high income countries such as the United States, Canada, Germany, and the United Kingdom (Supplementary Fig. 1). Information of funding sources from lower income countries was limited, with only one country (Greece) having a GDP below the top 50 of reported counties based on World Bank estimates [ 21 ]. Non-governmental organizations and local agencies made up a modest proportion of funding sources for the total of articles published (20%).

Author pronouns on climate change and infectious disease research. The self-identified pronouns of A first authors and B last (senior) authors of articles on climate change and disease from 2015 to 2020. The disparity between he/him pronoun usage over other pronouns was pronounced for senior authors. Authors’ pronoun usage in public settings may vary from their gender identities

Map of study locations by country. A The geographic representation of where studies were conducted (i.e., country where the data analyzed in the study originated) from 2015–2020 on climate change and infectious disease and B publications that fit the inclusion criteria as a proportion of human population in 2020 (per one million individuals). Population data were collected from the United Nations Population Division [ 26 ]. Darker color represents more publications conducted in or on the corresponding country. Grey indicates that no studies which fit the inclusion criteria were conducted in or on the corresponding countries. Shape file for map creation sourced from DIVA-GIS [ 84 , 85 ]

Map of lead author affiliation origins. The geographic representation of lead author affiliation origins for research on climate change and disease from 2015 to 2020. Darker color represents more publications originating from the corresponding country. Grey indicates that no studies which fit the inclusion criteria were conducted by authors affiliated with the corresponding countries. Blue points indicate the top ten publishing institutions globally for climate change and disease. Shape file for map creation sourced from DIVA-GIS [ 84 , 85 ]
Through this study we have revised the major trends in the current literature on climate change and infectious diseases. Our assessment identified both topical and geographic biases in the climate change and disease research arena. More specifically, we found that there was a notable focus on diseases which impact humans and upon arthropod-borne pathogens. Taxonomic bias, or the emphasis of study on specific organisms [ 27 ], has previously been identified in biodiversity and conservation science research [ 28 , 29 , 30 ]. Our results have identified taxonomic biases toward mammalian hosts and arthropod-borne pathogens and in climate change and infectious disease research. When certain taxa are over-represented in various scientific fields it is possible for them to draw both attention and funds away from less understood taxa [ 28 ]. It is possible that taxonomic bias has impacted the study of climate change and infectious disease by skewing research toward specific disease systems, suggesting an anthropocentric research approach potentially influenced by external forces, such as public health funding and disease burden [ 31 , 32 ]. Vector-borne diseases have considerable burden on human health, killing approximately 700,000 people annually [ 33 ]. A research emphasis on diseases affecting humans is, therefore, potentially unsurprising as human health is a driving force behind many research efforts and encompasses a large proportion of research and development funding [ 34 , 35 ]. Other research has shown that societal pressures correlate with taxonomic bias [ 28 ], which could explain why human-only and zoonotic diseases were so heavily studied as well.
Despite the anthropocentric nature of our results, many understudied taxa, such as amphibians, birds, and aquatic invertebrates, have higher risks of extinction due to infectious diseases than humans or other mammals [ 36 , 37 , 38 ]. Taxonomic bias in the study of infectious disease is concerning, as a lack of research effort could limit the understanding of diseases systems for threatened or endangered taxa. This in turn limits our capacities to understand how, where, and why diseases emerge in the wild. Risks of climate change impacts on lesser studied groups, such as wildlife and livestock, could still have public health effects due to spillover transmission of unknown pathogens [ 22 , 39 ]. The dearth of research on wildlife diseases could also lead to gaps of knowledge. Infectious diseases may harm ecological balance by reducing wildlife populations and decreasing overall biodiversity [ 40 , 41 , 42 ]. A large body of literature shows that ecological imbalances and biodiversity loss have detrimental effects on human health as well [ 39 , 43 , 44 , 45 ]. For instance, decreases in diversity of wildlife has been associated with increases risk of hantavirus spillover transmission from rodents to humans [ 46 , 47 , 48 , 49 ]. Public health efforts to study climate change and human health should consider biodiversity dimensions of spillover transmission for a more holistic ecosystem health approach.
We found that most lead authors were linked to higher-education institutions (i.e., universities or colleges), with fewer publications originating from governmental organizations or independent research institutions (Fig. 2 ). This bias towards academic-based research is not surprising considering that higher-education institutions often focus efforts on research and disseminating knowledge [ 50 ]. This result also indicates a poor active participation of stakeholders in governing bodies on climate change and health research, which could explain the slow progress of international policy on climate change and disease research. It is important to note, however, that most funding for the support of recent research publications originated from federal or national institutions (Additional file 1 : Fig. S1). While funding agencies constitute important stakeholders in the scientific publication process, agendas from funding sources may bias the research topics and discoveries reported [ 51 , 52 ]. For instance, publications with corporate funding are more likely to contribute to the polarization or politicization (i.e., contributing to the tension between political ideologies or identities) of climate change related topics [ 53 ]. We found that most articles reviewed for funding sources did not receive funding from corporate or industry agencies. Government funding is the main driver of science and provides research directions for non-government funding sources [ 52 ]. As such, an increase in government funding for climate change and infectious disease research accounting for environmental justice could transform the landscape of public and private research funding opportunities to reduce the inequities presented here. An increase in funding in the social science aspects of climate change may also facilitate the framing of climate change as a global social challenge, rather than a purely scientific endeavor with limited social legitimacy [ 54 ].
We also found that there was greater usage of he/him pronouns by lead and senior authors across the articles revised, suggesting that more male or male identified authors were present than female or female identified authors (Fig. 3 ). Gender discrepancies in authorship were more notable for senior authorship than for first authorship, which appears to be a general pattern in academic authorship inequity [ 55 ], even with increased authorship by women in recent decades [ 56 ]. Until recently, women or female-identified authors comprise a minority of researchers and trainees in science in general, which has resulted in authorship inequities that are expected to persist for some time [ 56 ]. Gender persistant inequity in authorship is specifically conerning within the field of climate change and infectious disease research due to its cross cutting social implications. Women are expected to experience greater climate change and health impacts as a result of their social and economic positions, and cultural discrimination [ 57 ]. As such it is important that women’s viewpoints and experiences are represented within the scientific literature to develop more effective and inclusive policies for climate change adaptation and mitigation.
In terms of geographic scale and location, we found that most climate change and infectious disease research was conducted at the regional and local scales (Fig. 2 ), suggesting that fine-scale studies dominate the field and our understanding of climate change impacts on human and animal health. Climate change and disease research also occurred principally in temperate areas (e.g., North America, Europe) rather than in tropical areas (e.g., sub-Saharan Africa, Latin America, and Pacific Southeast Asia) (Figs. 4 , 5 ). This spatial bias is present even when publications were corrected for country population. The research effort discrepancy between temperate vs tropical regions is concerning considering that tropical areas are the most at risk for emerging infectious diseases impacts [ 58 , 59 ]. Tropical areas are also experiencing drastic climate change effects, including reductions in food availability in short periods [ 60 ]. Tropical areas having limited to no climate change and disease research included Latin America, Northern and West Africa, and the Indo-pacific (Figs. 3 , 4 ). Furthermore, climate change is expected to increase the areas suitable for infectious agents in land and aquatic ecosystems [ 10 , 61 ]. For instance, the aquatic pathogen Vibrio cholerae , the causative agent of cholera, is expected to increase in regions where we found limited research effort [ 61 , 62 ]. Other areas which did not receive substantive research effort include extremely cold Arctic or Subarctic areas of Eurasia (Fig. 4 ). Permafrost regions such as these have recently experienced outbreaks of avian influenza (H5N1) [ 63 ], and previous reviews have identified melting permafrost as a reservoir of potentially viable and uncharacterized pathogens [ 64 ]. As such, a constituted effort to elucidated emerging infectious diseases in these regions should be undertaken to mitigate the risks of disease emergence. The confluence of susceptibility to both climate change impacts and infectious disease suggests a need for research in underrepresented areas reported here. Furthermore, underrepresentation of countries and human communities already disenfranchised and at greater risk for encountering infectious disease amplifies social inequity [ 7 ].
One caveat of our assessment is that publications from lower income or developing countries may not have been indexed in the publication data repositories accessed (i.e., Web of Science and PubMed) due to publication barriers such as language, publication fees, or lack of equitable partnerships or collaborative networks [ 65 , 66 , 67 , 68 , 69 ]. The potential misrepresentation of science from low-income countries highlights a possible equity issues within the dissemination of research which, in turn, could lead to the exclusion of relevant discoveries in the global health agenda [ 68 , 69 , 70 ]. A confirmation or publication bias could also be present in our results, as seen by the high number of papers which positively identified a climate change impact on infectious diseases. Previous research has commented on the scientific culture and potential dangers associated with the current emphasis on publishing only “significant” or “positive” results [ 71 , 72 , 73 , 74 ]. It is possible that researchers were reluctant or unable to publish negative or inconclusive results, thus skewing the conclusions of this sample. Furthermore, while we found that many articles either found a definitive climate change impact, or concluded that climate change could “possibly” or “potentially” impact the disease system being assessed, these findings were based upon the author’s interpretation of their results and may be an exaggerated interpretation of the data. Finally, while we sought to identify the distribution of authorship via author pronoun usage, there could be discrepancies present between the pronouns publicly available for the authors and the gender identities they have privately. This discrepancy is to be expected considering the discriminatory practices in academia against lesbian, gay, bisexual, transgender, and queer (LGBTQ+) scientists [ 75 , 76 , 77 ].
We found that both geographic and taxonomic trends were present in recent studies assessing climate change and the burden of infectious disease. The majority of research was focused on vector-borne pathogens and was conducted in well-developed, high-income countries with temperate climates, neglecting directly-transmitted diseases in tropical regions. The anthropocentric signal in research effort may contribute to a lack of understanding of climate change effects on wildlife systems. The underrepresentation of some taxonomic groups of pathogens and hosts, pathogen transmission types, and geographic areas should be of global health concern, as areas and diseases neglected may become sources of emerging zoonotic diseases. An ecosystem-based framework to study disease responses to climate change could mitigate topical and taxonomic biases identified here. Viral zoonoses outbreaks at the local level in underrepresented countries such as Madagascar, Saudi Arabia, and Indonesia have led to prolific human epidemics of plague, Middle East respiratory syndrome, and cholera in recent years [ 78 ], highlighting the need for more research in regions underrepresented in the literature. The recent coronavirus disease pandemic also highlights the need for more research on directly transmitted pathogens circulating in wildlife [ 79 ]. Furthermore, research is still needed to understand the linkages between patterns of research funding with climate change and infectious disease studies. Understanding the funding landscape (e.g., agencies prioritizing certain regions, diseases, and topics) could further elucidate the relationship between research bias, research equity, and funding allocation.
The impact of climate change research on intergovernmental policy and vice versa is both tractable and increasingly important [ 80 , 81 ]. Policy changes to address the biases presented here, including the diseases studied, areas, and identities of leading authors, should be prioritized by both funding agencies and the scientific community. Policy change could include, for example, the prioritization of infectious disease research and surveillance at the human-wildlife interface within the context of climate change, funding prioritizing scientists from minority groups, and neglected geographic regions. Addressing research inequity will help build human capacity, surveillance, and scientific infrastructure to better prepare and strengthen the global health response to climate change threats [ 82 ]. Furthermore, research foundations in high-income countries should implement and maintain inclusive-collaboration practices to value contributions by local scientists in countries underrepresented in this review to advance research equity as a means towards effective prevention of future emerging diseases from their sources. Building political and social support behind climate change and infectious disease research will be essential under the expected rates of climatic variation in the near future [ 83 ]. In conclusion, there is an urgent need to increase research effort for neglected disease systems and geographies, and there is a need to re-examine aspects of environmental justice from the scientists leading these studies to the local beneficiaries for the advancement of infectious diseases research in the context of climate change.
Availability of data and materials
Not applicable.
Abbreviations
Gross domestic product
IPCC. Climate Change 2014 Part A: Global and Sectoral Aspects. Field CB, Barros VR, Dokken DJ, Mach KJ, Mastrandrea MD, Chatterjee TEBM, et al. New York: Cambridge University Press; 2014. p. 169–833
IPCC. Climate Change 2014 impacts, adaptation, and vulnerability Part B: Regional aspects: Working group ii Contribution to the fifth assessment report of the intergovernmental panel on climate change. Barros VR, Field CB, Dokken DJ, Mastrandrea MD, Mach KJ, Chatterjee M, et al. New Yorka: Cambridge University Press; 2014. p. 1133–1655.
Altizer S, Ostfeld RS, Johnson PTJ, Kutz S, Harvell CD. Climate change and infectious diseases: from evidence to a predictive framework. Science. 2013;341:514–9.
Article CAS PubMed Google Scholar
Vora N. Impact of anthropogenic environmental alterations on vector-borne diseases. Medscape Gen Med. 2008;10:238.
Google Scholar
Price SJ, Leung WTM, Owen CJ, Puschendorf R, Sergeant C, Cunningham AA, et al. Effects of historic and projected climate change on the range and impacts of an emerging wildlife disease. Glob Chang Biol. 2019;25:2648–60.
Article PubMed Google Scholar
Caminade C, McIntyre KM, Jones AE. Impact of recent and future climate change on vector-borne diseases. Ann N Y Acad Sci. 2019;1436:157–73.
Anwar A, Anwar S, Ayub M, Nawaz F, Hyder S, Khan N, et al. Climate change and infectious diseases: evidence from highly vulnerable countries. Iran J Public Health. 2019;48:2187–95.
PubMed PubMed Central Google Scholar
Ryan SJ, Carlson CJ, Mordecai EA, Johnson LR. Global expansion and redistribution of Aedes -borne virus transmission risk with climate change. PLoS Negl Trop Dis. 2018;13: e0007213.
Article Google Scholar
Messina JP, Brady OJ, Golding N, Kraemer MUG, Wint GRW, Ray SE, et al. The current and future global distribution and population at risk of dengue. Nat Microbiol. 2019;4:1508–15.
Article CAS PubMed PubMed Central Google Scholar
Siraj AS, Santos-Vega M, Bouma MJ, Yadeta D, Carrascal DR, Pascual M. Altitudinal changes in malaria incidence in highlands of Ethiopia and Colombia. Science. 2014;343:1154–9.
Peterson J, Assistant RN, Pearce PF, Associate F, Ferguson LA, Associate F, et al. Understanding scoping reviews: definition, purpose, and process. J Am Assoc Nurse Pract. 2017;29:12–6.
Munn Z, Peters MDJ, Stern C, Tufanaru C, McArthur A, Aromataris E. Systematic review or scoping review? Guidance for authors when choosing between a systematic or scoping review approach Zachary. BMC Med Res Methodol. 2018;18:143.
Article PubMed PubMed Central Google Scholar
Levac D, Colquhoun H, O’Brien KK. Scoping studies: advancing the methodology. Implement Sci. 2010;5:6–9.
Arksey H, O’Malley L. Scoping studies: towards a methodological framework. Int J Soc Res Methodol Theory Pract. 2005;8:19–32.
McCallum H. Models for managing wildlife disease. Parasitology. 2016;143:805–20.
Taylor LH, Latham SM, Woolhouse MEJ. Risk factors for human disease emergence. Philos Trans R Soc B Biol Sci. 2001;356:983–9.
Article CAS Google Scholar
Marcial LH, Hemminger BM. Scientific data repositories on the Web: An initial survey. J Am Soc Inf Sci Technol. 2010;61:2029–48.
McEntyre J, Lipman D. PubMed: bridging the information gap. Can Med Assoc J. 2001;164:1317–9.
CAS Google Scholar
Jones L. Language and gender identities. In: Preece S, editor. Routledge handb lang identity. New York: Routledge; 2016. p. 210–24.
Gray J. Language and non-normative sexual identities. In: Preece S, editor. Routledge handb lang identity. New York: Routledge; 2016. p. 225–40.
The World Bank. Gross Domestic Product 2021. https://databankfiles.worldbank.org/public/ddpext_download/GDP.pdf . Accessed 12 April 2023.
Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990–3.
ESRI. ArcGIS Desktop 10.5. Redlands, CA: Environmental Systems Research Institute; 2019.
Wickham H, RStudio. tidyverse: Easily install and load the “Tidyverse.” New York: Springer-Verlag; 2018.
Bivand R, Lewin-Koh N. maptools: Tools for handling spatial objects. R package version 0.9.8. 2019.
United Nations Department of Economic and Social Affairs Population Division. United Nations World Population Prospects. https://population.un.org/wpp/ . Accessed 12 January 2021.
Bonnet X, Shine R, Lourdais O. Taxonomic chauvinism. Trends Ecol Evol. 2002;17:1–3.
Troudet J, Grandcolas P, Blin A, Vignes-Lebbe R, Legendre F. Taxonomic bias in biodiversity data and societal preferences. Sci Rep. 2017;7:1–14.
Clark JA, May RM. Taxonomic bias in conservation research. Science. 2002;297:191–2.
Lawler JJ, Aukema JE, Grant JB, Halpern BS, Kareiva P, Nelson CR, et al. Conservation science: a 20-year report card. Front Ecol Environ. 2006;4:473–80.
Gillum LA, Gouveia C, Dorsey ER, Pletcher M, Mathers CD, McCulloch CE, et al. NIH disease funding levels and burden of disease. PLoS ONE. 2011;6: e16837.
Gross CP, Anderson GF, Powe NR. The relation between funding by the National Institutes of Health and the burden of disease. N Engl J Med. 1999;340:1881–7.
World Health Organizations report: Vector-borne diseases. https://www.who.int/news-room/fact-sheets/detail/vector-borne-diseases . Accessed 10 September 2022.
United States Congressional Research Service. U.S. Research and Development Funding and Performance: Fact Sheet R44307 https://crsreports.congress.gov . Accessed 1 Octobaer 2022.
United States National Institues of Health. Budget for the National Institues of Health (NIH): History of NIH Appropriations. https://www.nih.gov/about-nih/what-we-do/budge . Accessed 1 October 2022.
McKinney ML. High rates of extinction and threat in poorly studied taxa. Conserv Biol. 1999;13:1273–81.
Thomas CD, Cameron A, Green RE, Bakkenes M, Beaumont LJ, Collingham YC, et al. Extinction risk from climate change. Nature. 2004;427:145–8.
Uthicke S, Momigliano P, Fabricius KE. High risk of extinction of benthic foraminifera in this century due to ocean acidification. Sci Rep. 2013;3:1769.
Article PubMed Central Google Scholar
Gibb R, Redding DW, Chin KQ, Donnelly CA, Blackburn TM, Newbold T, et al. Zoonotic host diversity increases in human-dominated ecosystems. Nature. 2020;584:398–402.
Scheele BC, Pasmans F, Skerratt L, Berger L, Martel A, Beukema W, et al. Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science. 2019;363:1459–63.
LaDeau SL, Kilpatrick AM, Marra PP. West Nile virus emergence and large-scale declines of North American bird populations. Nature. 2007;447:710–3.
Dadam D, Robinson RA, Clements A, Peach WJ, Bennett M, Rowcliffe JM, et al. Avian malaria-mediated population decline of a widespread iconic bird species. R Soc Open Sci. 2019;6: 182197.
Keesing F, Ostfeld RS. Impacts of biodiversity and biodiversity loss on zoonotic diseases. Proc Natl Acad Sci USA. 2021;118: e2023540118.
Johnson CK, Hitchens PL, Pandit PS, Rushmore J, Evans TS, Young CCW, et al. Global shifts in mammalian population trends reveal key predictors of virus spillover risk. Proc R Soc B Biol Sci. 2020;287: e20192736.
Díaz S, Fargione J, Chapin FS, Tilman D. Biodiversity loss threatens human well-being. PLoS Biol. 2006;4:1300–5.
Ruedas LA, Salazar-Bravo J, Tinnin DS, Armién B, Cáceres L, García A, et al. Community ecology of small mammal populations in Panamá following an outbreak of Hantavirus pulmonary syndrome. J Vector Ecol. 2004;29:177–91.
PubMed Google Scholar
Keesing F, Belden LK, Daszak P, Dobson A, Harvell CD, Holt RD, et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature. 2010;468:647–52.
Tian H, Stenseth NC. The ecological dynamics of hantavirus diseases: From environmental variability to disease prevention largely based on data from China. PLoS Negl Trop Dis. 2019;13:1–19.
Clay CA, Lehmer EM, St. Jeor S, Dearing MD. Sin Nombre virus and rodent species diversity: a test of the dilution and amplification hypotheses. PLoS ONE. 2009;4:e6467.
Amado Mateus M, Juarez AF. Reputation in higher education: a systematic review. Front Educ. 2022;7: 925117.
Jacob BA, Lefgren L. The impact of research grant funding on scientific productivity. J Public Econ. 2011;95:1168–77.
Lanahan L, Graddy-Reed A, Feldman MP. The domino effects of federal research funding. PLoS ONE. 2016;11: e0157325.
Farrell J. Corporate funding and ideological polarization about climate change. Proc Natl Acad Sci USA. 2016;113:92–7.
Overland I, Sovacool BK. The misallocation of climate research funding. Energy Res Soc Sci. 2020;62: 101349.
Broderick NA, Casadevall A. Gender inequalities among authors who contributed equally. Elife. 2019;8: e36399.
Holman L, Stuart-Fox D, Hauser CE. The gender gap in science: How long until women are equally represented? PLoS Biol. 2018;16: e2004956.
Sellers S. Gender and climate change: a closer look at existing evidence global. Glob Gend Clim Alliance. 2016;27:9–31.
Allen T, Murray KA, Zambrana-torrelio C, Morse SS, Rondinini C, Di Marco M, et al. Global hotspots and correlates of emerging zoonotic diseases. Nat Commun. 2017;8:923–33.
Morse SS, Mazet JAK, Woolhouse M, Parrish CR, Carroll D, Karesh WB, et al. Prediction and prevention of the next pandemic zoonosis. Lancet. 2012;380:1956–65.
Wheeler T, von Braun J. Climate change impacts on global food security. Science. 2013;341:508–13.
Vezzulli L, Grande C, Reid PC, Hélaouët P, Edwards M, Höfle MG, et al. Climate influence on Vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc Natl Acad Sci USA. 2016;113:e5062–71.
Escobar LE, Ryan SJ, Stewart-Ibarra AM, Finkelstein JL, King CA, Qiao H, et al. A global map of suitability for coastal Vibrio cholerae under current and future climate conditions. Acta Trop. 2015;149:202–11.
Canavan BC. Opening Pandora’s Box at the roof of the world: landscape, climate and avian influenza (H5N1). Acta Trop. 2019;196:93–101.
Wu R, Trubl G, Taş N, Jansson JK. Permafrost as a potential pathogen reservoir. One Earth. 2022;5:351–60.
Konno K, Akasaka M, Koshida C, Katayama N, Osada N, Spake R, et al. Ignoring non-English-language studies may bias ecological meta-analyses. Ecol Evol. 2020;10:6373–84.
National Science Board. Publications Output: U.S. Trends and International Comparisons. Sci Eng Indic. 2022. 2021.
Gazni A, Sugimoto CR, Didegah F. Mapping world scientific collaboration: authors, institutions, anc countries. J Am Soc Inf Sci Technol. 2012;63:323–35.
Shumba CS, Lusambili AM. Not enough traction: barriers that aspiring researchers from low- and middle-income countries face in global health research. J Glob Heal Econ Policy. 2021;1: e2021002.
Boum Y, Burns BF, Siedner M, Mburu Y, Bukusi E, Haberer JE. Advancing equitable global health research partnerships in Africa. BMJ Glob Heal. 2018;3:5–8.
Dimitris MC, Gittings M, King NB. How global is global health research? A large-scale analysis of trends in authorship. BMJ Glob Heal. 2021;6: e003758.
Barnett AG, Wren JD. Examination of CIs in health and medical journals from 1976 to 2019: an observational study. BMJ Open. 2019;9: e032506.
Emerson GB, Warme WJ, Wolf FM, Heckman JD, Brand RA, Leopold SS. Testing for the presence of positive-outcome bias in peer review: a randomized controlled trial. Arch Int Med. 2010;170:1934–9.
Fanelli D. Negative results are disappearing from most disciplines and countries. Scientometrics. 2012;90:891–904.
Head ML, Holman L, Lanfear R, Kahn AT, Jennions MD. The extent and consequences of P-hacking in science. PLoS Biol. 2015;13: e1002106.
LaSala MC, Jenkins DA, Wheeler DP, Fredriksen-Goldsen KI. LGBT faculty, research, and researchers: risks and rewards. J Gay Lesbian Soc Serv. 2008;20:253–67.
Gibney E. Discrimination drives LGBT + scientists to think about quitting strange materials excite physicists. Nature. 2019;571:16–7.
Dyer J, Townsend A, Kanani S, Matthews P, Palermo A. Exploring the workplace for LGBT+ physical scientists. R Soc Chem Inst Phys R Astron Soc. 2019;1:10–42.
Piret J, Boivin G. Pandemics throughout history. Front Microbiol. 2021;11: e631736.
Beyer RM, Manica A, Mora C. Shifts in global bat diversity suggest a possible role of climate change in the emergence of SARS-CoV-1 and SARS-CoV-2. Sci Total Environ. 2021;767: e145413.
Bornmann L, Haunschild R, Boyack K, Marx W, Minx JC. How relevant is climate change research for climate change policy? An empirical analysis based on Overton data. PLoS ONE. 2022;17: e0274693.
Campbell-Lendrum D, Manga L, Bagayoko M, Sommerfeld J. Climate change and vector-borne diseases: What are the implications for public health research and policy? Philos Trans R Soc B Biol Sci. 2015;370:20130552.
Hess J, Boodram LLG, Paz S, Stewart Ibarra AM, Wasserheit JN, Lowe R. Strengthening the global response to climate change and infectious disease threats. BMJ. 2020;371: m3081.
Baker RE, Mahmud AS, Miller IF, Rajeev M, Rasambainarivo F, Rice BL, et al. Infectious disease in an era of global change. Nat Rev Microbiol. Springer US; 2022;20:193–205.
Hijmans RJ. DIVA-GIS (2017) Free Spatial Data by Country. http://www.diva-gis.org/gdata . Accessed 12 October 2019.
Hijmans RJ, Guarino L, Cruz M, Rojas E. Computer tools for spatial analysis of plant genetic resources data: 1. DIVA-GIS. Plant Genet Resour Newsl. 2001;127:15–9.
Download references
Acknowledgements
Authors thank Sarah M. Karpanty, Mark Ford, Steven N. Winter, Mariana Castaneda-Guzman, Diego Soler-Tovar, Caroline Ilse, Abigail Parch, Tabatha Gentry, David Treanor, Alma Talcott, and Victor Jose Catalan who contributed greatly to the completion of this work.
This study was supported by the National Science Foundation award: Human–Environment and Geographical Sciences Program (2116748), the Institute for Critical Technology and Applied Science, Virginia Tech: ICTAS-JFP-2022-2023 program, and the Virginia Tech College of Natural Resources and Environment Environmental Security Grant program.
Author information
Authors and affiliations.
Virginia Tech Graduate School, Translational Biology, Medicine, and Health Program, Blacksburg, VA, USA
Paige Van de Vuurst
Department of Fish and Wildlife Conservation, Virginia Tech, Blacksburg, VA, USA
Paige Van de Vuurst & Luis E. Escobar
Center for Emerging Zoonotic and Arthropod-Borne Pathogens, Virginia Tech, Blacksburg, VA, USA
Global Change Center, Virginia Tech, Blacksburg, VA, USA
Luis E. Escobar
Facultad de Ciencias Agropecuarias, Universidad de La Salle, Bogotá, Colombia
You can also search for this author in PubMed Google Scholar
Contributions
Both authors designed and wrote the first draft of this commentary. All authors contributed to the development, review, and approval of the last version of this article. All authors read and approved the final manuscript.
Corresponding author
Correspondence to Luis E. Escobar .
Ethics declarations
Ethics approval and consent to participate, consent for publication.
Consent was obtained from Luis E. Escobar for the publication of any data of images herein.
Competing interests
The authors declare that there are no competing interests.
Supplementary Information
Additional file 1: figure s1..
Sources of publication funding or support from articles published in 2020. For all articles which fit the inclusion criteria that were published in the year 2020 we extracted the funding or support source listed in the article. These funding sources were classified as federal, non-governmental organizations such as charities or independent research organizations, local agencies, or a combination of federal and academic or federal and industry support.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Van de Vuurst, P., Escobar, L.E. Climate change and infectious disease: a review of evidence and research trends. Infect Dis Poverty 12 , 51 (2023). https://doi.org/10.1186/s40249-023-01102-2
Download citation
Received : 10 January 2023
Accepted : 04 May 2023
Published : 16 May 2023
DOI : https://doi.org/10.1186/s40249-023-01102-2
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Climate change
- Infectious disease
- Research trend
- Systematic review
Infectious Diseases of Poverty
ISSN: 2049-9957
- Submission enquiries: Access here and click Contact Us
- General enquiries: [email protected]
Hepatitis E virus immunosuppressed animal models
- Open access
- Published: 12 September 2024
- Volume 24 , article number 965 , ( 2024 )
Cite this article
You have full access to this open access article

- Kush Kumar Yadav 1 , 2 &
- Scott P. Kenney 1 , 2
1 Altmetric
Hepatitis E virus (HEV) is an important emerging pathogen producing significant morbidity in immunosuppressed patients. HEV has been detrimental to solid organ transplant (SOT) patients, cancer patients, and HIV-positive patients, where chronic HEV infections occur. Blood-borne transfusions and multiple cases of chronic HEV infection in transplant patients have been reported in the past few decades, necessitating research on HEV pathogenesis using immunosuppressed animal models. Numerous animal species with unique naturally occurring HEV strains have been found, several of which have the potential to spread to humans and to serve as pathogenesis models. Host immunosuppression leads to viral persistence and chronic HEV infection allows for genetic adaptation to the human host creating new strains with worse disease outcomes. Procedures necessary for SOT often entail blood transfusions placing immunosuppressive patients into a “high risk group” for HEV infection. This scenario requires an appropriate immunosuppressive animal model to understand disease patterns in these patients. Hence, this article reviews the recent advances in the immunosuppressed animal models for chronic HEV infection with emphasis on pathogenesis, immune correlates, and the liver pathology associated with the chronic HEV infections.
Similar content being viewed by others

New Animal Models for Hepatitis C

Dual Reconstituted Mice for Hepatotropic Pathogens

A Chimeric Mouse Model to Study Immunopathogenesis of HCV Infection
Avoid common mistakes on your manuscript.
Hepatitis E virus (HEV) is an interesting topic in the field of emerging infectious diseases. HEV ranks 6th on a list of spillover viruses with significant health risks to humans [ 1 ]. HEV causes both acute and chronic infection in humans and is the leading cause of acute viral gastroenteritis worldwide [ 2 ]. Chronic HEV infection has been reported in patients receiving a solid organ transplant (SOT) [ 3 ], with blood disorders [ 4 ], and in human immunodeficiency virus (HIV)-positive patients [ 5 ]. A sustained rise in liver enzyme levels and viral RNA detection in the blood and feces for at least six months following the acute phase of infection are considered indicators of a chronic HEV infection [ 3 ]. Chronic HEV infection is becoming more common, especially in patients receiving SOTs who require long term immunosuppression to prevent organ rejection [ 6 , 7 ].
HEV is a non-enveloped, positive-sense RNA virus approximately 27–34 nm in diameter and contains a 6.4–7.2 kb viral genome encoding three primary open reading frames (ORFs). HEV is a member of the family Hepeviridae comprised of two subfamilies, five genera, and at least ten species [ 8 ]. Within the Hepeviridae, the Orthohepevirinae subfamily has several genera known to infect mammalian species. To date only Paslahepevirus genus members (previously known as Orthohepevirus A ) and some members of the Rocahepevirus (previously known as Orthohepevirus C ) genus have been found to infect immunosuppressed humans. More specifically, Paslahepevirus balayani genotype (gt)3, gt4, gt7, and Rocahepevirus ratti gt1 have been reported to infect immunosuppressed patients [ 3 , 4 , 5 , 9 , 10 , 11 , 12 , 13 ].
A dearth of robust and tractable animal and cell culture models that accurately and fully mimic hepatitis E disease as observed in humans has made studying the pathogenesis of HEV a difficult undertaking. For instance, although the fecal-oral pathway is the main means of HEV transmission, the exact mechanism by which the virus particles move from the gastrointestinal system to the liver remained mostly unknown. Recently, clinical samples from a patient with a chronic HEV infection were shown to have intestinal crypts containing HEV RNA and ORF2 antigens leading to crypt cells being tested and confirmed as supporting HEV replication in vitro [ 14 ]. Animal models in which the immune response can be modified are necessary to uncover nuances of the HEV lifecycle that are otherwise suppressed and unobservable during natural infection. Furthermore, most HEV infections in immunocompromised individuals tend to become chronic infections [ 15 ] resulting in differential and prolonged pathogenesis compared to immunocompetent hosts. These persistent HEV infections can result in nodules, fibrotic remodeling, and eventually cirrhosis in the liver [ 16 ]. Consequently, it is imperative to comprehend the consequences of hepatitis E in individuals with impaired immune systems, necessitating the use of reliable and physiologically applicable animal models.
Here we have delineated HEV pathological features in humans resulting from chronic HEV infections during generalized immunosuppression. This information is intended to help readers understand the need for an appropriate immunosuppressive animal model for HEV. We discuss the pathogenesis of HEV in immunosuppressed cynomolgus monkeys, pigs, rabbits, mice, rats, and Mongolian gerbils while comparing the pathology, immune correlates, and drug screening between animal models that produce chronic HEV infection.
Chronic HEV infection pathology in humans
Persistent HEV replication for at least six months, moderate increase of liver enzymes, and an infrequent correlation with clinical indications commonly seen in immunosuppressed patients are characteristics of chronic human HEV infections [ 17 ]. Generally, gt1 and gt2 Paslahepevirus balyani strains are not attributed to chronic HEV infections whereas gt3 and gt4 are much more frequently associated with chronic infections making these strains a priority for animal infection models. The majority of otherwise healthy patients with P balayani gt3 or gt4 infection have no symptoms, but these viruses can have catastrophic consequences for immunocompromised or immunodeficient individuals [ 15 ]. Developed nations are seeing an increase in P balayani gt3 or gt4 infections, and immunocompromised persons are particularly vulnerable to persistent HEV-related liver fibrosis. Recently, chronic HEV infection of immunosuppressed patients has been reported to be caused by Rocahepevirus ratti gt1, despite the fact that this HEV strain significantly differs genetically from P balayani HEV strains [ 9 ].
In real-world clinical situations, chronic HEV infection develops in approximately 60% of transplant patients who have preexisting HEV infections or are exposed during transplantation via transfusion with 10% of these chronic patients developing cirrhosis [ 18 , 19 ]. When hepatitis E is diagnosed in SOT patients undergoing tacrolimus medication, the risk is elevated [ 18 ]. Following SOT, reports have been made regarding the incidence of a de novo HEV infection as well as the risk of reinfection in the patients suggesting this may occur in 1 to 1.5% of patients [ 20 , 21 ]. Interestingly, a case of chronic HEV gt3 has been reported in a pregnant woman who was under immunosuppressive agents for ulcerative colitis [ 22 ]. Even though the chronicity was observed, no adverse event during pregnancy was reported and resolution after delivery was without complications [ 22 ]. Another case of chronic HEV in pregnancy was reported in a kidney transplant patient. HEV gt3 infection was diagnosed during the first trimester of pregnancy [ 23 ]. Interestingly, HEV RNA increased by more than 1.5 log factor at the beginning of the third trimester but after the Cesarean section at the 38th week of gestation, the newborn and the placenta tested negative for HEV RNA. HEV was not detectable in the mother either in serum or stool even after six months [ 23 ] suggesting the role of pregnancy in HEV sustainability. A recent report of a pregnant woman who had undergone liver transplantation demonstrated the presence of HEV gt3 infection at the 34th week of gestation [ 24 ]. Placenta and breast milk tested positive for HEV RNA [ 24 ]. However, the newborn did not demonstrate HEV RNA either in serum or stool (tested at 1–5, 15th days and 1 month) and was negative for anti-HEV IgM but was positive for anti-HEV IgG [ 24 ]. In contrast to the previous case, HEV did not clear even after 3 months of delivery and thus, the mother was treated with the ribavirin for 16 months until the HEV RNA was undetectable in the serum and stool [ 24 ].
Clinical research has shown that for treatment plan guidance fecal HEV shedding is a more accurate indicator for relapse prediction in chronic HEV infection than viremia [ 25 , 26 , 27 ]. Nonetheless, it has been shown that immunosuppressed individuals’ blood and urine contain substantial concentrations of HEV antigens [ 28 , 29 ]. Liver fibrosis and cirrhosis can develop quickly in SOT patients who have a persistent HEV infection [ 18 , 30 ]. As expected, overexpression of host genes implicated in fibrogenesis was also discovered by RNA sequencing of host transcripts in fibrotic liver tissues [ 31 ].
According to histopathological findings, lymphocytic portal infiltrates with piecemeal necrosis (interface hepatitis) have been observed in HEV-infected heart and liver transplant recipients [ 3 , 20 , 32 , 33 ]. Patients with SOT who progressed to chronic infection have low peak levels of ALT (Alanine aminotransferases) and AST (Aspartate aminotransferases) [ 18 ]. In addition to SOT patients, rheumatological patients, hematological patients, HIV-infected patients, and hematopoietic stem cell transplant (HSCT) patients are in the high-risk group to develop severe chronic HEV infection [ 34 , 35 ]. Furthermore, tacrolimus usage during HEV infection has been linked to immune-mediated and drug-induced severe thrombocytopenia [ 36 , 37 ]. Moreover, neurological conditions, renal damage, severe pancreatitis, and hematological abnormalities are extrahepatic symptoms of chronic HEV infection [ 33 ]. To help the reader comprehend the pathogenicity of chronic HEV infection and the need for a suitable immunosuppressed animal model, immunosuppressed chronic HEV infection has been summarized (Table 1 ).
Ideal immunosuppressive animal model attributes
Replicating the exact clinical disease and pathology of chronic human HEV infection in an animal model is extremely difficult. For in-depth HEV research, the ideal immunosuppressive animal model would include all the following traits.
Develops liver specific pathology such as fibrosis and cirrhosis with alteration in liver enzymes (ALT, AST, and gamma glutamyl transferases (GGT)).
Immunosuppressive drugs used in humans should be effective in lowering the immune response in the animal model.
Animals should be susceptible to human derived HEV strains.
Immunological responses to HEV should mimic humans.
Chronic HEV infection in the immunosuppressed animal should mimic the prolonged viremia and fecal viral shedding seen in humans.
Animal organ anatomical structure and physiological function should be close to humans.
Experimental tool kits and reagents should be readily available for the animal model.
Animal models should be genetically manipulatable such that knock-in or knock-out studies can be conducted.
Immunosuppressed animal models for studying chronic HEV infection
To obtain precise and accurate results, it is imperative to consistently replicate the human disease state in an animal model. An important factor to consider when selecting an appropriate HEV model is the animal’s susceptibility to a natural HEV infection. Moreover, using a particular animal species as a model for HEV infectious disease requires careful evaluation of the species’ anatomical, physiological, genetic, and biochemical parallels to humans.
Uncertainty surrounds the dynamics of HEV adaptation and propagation within genotypes that permit host range expansion. Numerous animal species have been found to have unique circulating HEV strains since the first animal HEV was discovered in pigs in 1997, followed by the first bird strain in 1998, the first rabbit strain in 2009, and the first rat strain in 2010. Several of these strains have the potential to spread to humans and cause chronic HEV infection. In order to study the outcomes of persistent hepatitis E in immunocompromised people, current immunocompromised animal models that replicate chronic HEV have been summarized below (Table 2 ).
Cynomolgus monkeys
When assessing the zoonotic potential of HEV, non-human primates (NHPs) are frequently the most appropriate model. The principal models used to research the clinical course of HEV infection are nonhuman primates (NHP), such as Macaca fascicularis (cynomolgus monkeys) and Macaca mulatta (rhesus monkeys). Despite not being HEV’s natural host, NHPs are vulnerable to experimental infection with P balayani gt1, gt2, gt3, and gt4 [ 56 , 57 , 58 , 59 ]. Cynomolgus monkeys were the first NHPs employed in experimental HEV infection research [ 60 ]. Additionally, a wide range of immunosuppressive drugs, including tacrolimus, have been developed in preclinical trials using cynomolgus monkeys, which are thought to be an excellent model for human organ transplantation [ 61 , 62 ].
Experimental infection of cynomolgus monkeys under tacrolimus treatment and concurrent infection with P balayani gt3 Brazilian strain mimicked chronic HEV infection in humans. Chronic HEV signs such as persistent viremia, fecal viral shedding, and elevated liver enzymes, with gross and microscopic hepatitis observed in the liver (Fig. 1 ) [ 46 ].

Summary of immunosuppressed cynomolgus monkey model with intravenous (IV) HEV inoculation. HEV gt3 was used for the study. Interestingly, chronic hepatitis was evident but with the absence of fibrosis and cirrhosis in liver
Briefly, out of 4 NHPs used in the study, 3 of them became chronically infected. The clinical data presented during the acute phase of infection such as liver enzymes and antibody titers failed to predict chronicity in the monkeys. It’s interesting to note that three of the monkeys that had chronic infections showed signs of a very delayed seroconversion and a prolonged elevation in liver enzymes like ALT and AST [ 46 ]. Abnormal fat deposits in the liver parenchyma were directly correlated with decreased cholesterol in the plasma of chronically infected monkeys. There was no significant difference in the white blood cell (WBC) and platelet count due to the immunosuppressive drugs used in the monkeys before and after the infection [ 46 ].
Active replication in the liver and associated tissues such as gall bladder, spleen, and pancreas were demonstrated by the presence of HEV negative-stranded RNA [ 46 ]. Liver histopathology reported hepatocellular ballooning, scattered apoptosis, and lobular focal inflammation with microscopic necrotic features. After 4 months, immunosuppressed and HEV infected monkeys progressed to chronic hepatitis but there were no signs of liver fibrosis. Type 2 diabetes mellitus was reported in one of the monkeys under immunosuppression and HEV infection resolved after the discontinuation of the immunosuppressive drug [ 46 ].
Despite the fact that NHPs have been used in several studies, their inability to function as a natural host of HEV and the lack of liver fibrosis render them as a less suitable model for researching chronic HEV infection in humans. Furthermore, with chimpanzees and other great apes now subject to stringent restrictions on research, the use of these animals in future research is also constrained due to cost and ethical concerns with invasive biomedical research in primates.
Pigs are the most studied source for xenotransplantation in humans [ 63 ]. Multiple immunosuppressive drugs used in humans during SOT have been studied in pigs [ 64 , 65 ]. Pigs are anatomically, physiologically, and immunologically similar to humans and thus are a potentially good model for mimicking chronic hepatitis E in humans, particularly SOT patients [ 66 , 67 ].
Zoonotic P balayani gt3 is responsible for many of the cases in humans leading to the development of chronic hepatitis [ 68 , 69 ]. Pigs are a known reservoir for P balayani gt3 and gt4, allowing pigs to be used as a natural host model for pathogenesis and therapeutic studies [ 70 ].
Mycophenolate mofetil and tacrolimus are the most commonly used drugs in SOT patients. Experimental chronic HEV infection in pigs mimicked humans by utilizing a combinatorial approach of drugs and the human HEV P balayani gt3 (US2 strain) (Fig. 2 ) [ 67 ].

Summary of immunosuppressed pig model with intravenous (IV) HEV inoculation. Th1, Th2 cytokines and CD4 + T cells were reduced during the acute phase, however, CD8 + T cells increased during the chronic phase of infection
Chronic HEV infection was produced in pigs as fecal viral shedding was seen for at least 5–14 weeks longer in comparison to the immunocompetent pigs [ 67 ]. On the other hand, viremia failed to be a predictor for chronic HEV infection demonstrating equal viral titers to immunocompetent pigs. Histopathology reports did not show any significant differences in the liver of immunocompetent infected pigs compared to chronically infected pigs [ 67 ]. The immunosuppressive drug treated and infected group showed reduction in Th1 cytokines (IL-2 and IL-12) and no significant changes in the IFN-γ cytokine levels in blood when compared to the immunocompetent and infected group. However, the CD4 + CD8 + T cell activation was decreased in the drug treated and infected group. During the chronic phase of infection, CD4 + T cells producing IL-4 increased in the blood of the immunosuppressed and infected pigs. A clear disease progression was evident by the shift of the immune response from CD4 + CD8 − T-cell population to CD4 + CD8 + T-cell population in the immunosuppressed pigs [ 67 ].
Another study utilized tacrolimus-based regimen demonstrating persistent viremia for 11 weeks post inoculation [ 48 ]. Liver inflammation and fibrosis were reported in the immunosuppressed pigs [ 48 ]. They revealed a unique compartmentalization of HEV genomes in the feces and intestinal tissues, supporting extrahepatic replication in the digestive tract [ 48 ].
Pigs only develop subclinical infection when infected with HEV thus the immunological response in symptomatic human HEV patients cannot be mimicked in pigs. The absence of liver fibrosis and decreased frequencies of HEV-specific T-cells in peripheral blood indicate that pigs are not the most suitable model to fully replicate the chronic HEV patient condition in humans. Long term dosing of pigs with immunosuppressive drugs may also be cost prohibitive due to their propensity for high weight gain.
Rabbits are a well-studied model for renal transplantation [ 71 ], stem cell therapy [ 72 ], and drug based immunosuppression studies [ 73 ]. Phylogenetically, rabbits are more closely related to primates than to rodents [ 74 ]. Zoonotic P balayani gt4 strains and rabbit specific strains (gt3) are capable of infecting rabbits [ 75 , 76 ]. Extrahepatic replication of HEV in the brain, heart, lung, kidney, spleen, and placenta has been demonstrated in SPF rabbits [ 77 , 78 , 79 ]. Easy availability of rabbits and minimum handling requirements while mimicking extrahepatic tissue lesions seen in humans make them a good model. Furthermore, rabbits and humans share similarities in airway anatomy and inflammatory responses [ 80 , 81 ]. The size of rabbits enables non-lethal observation of physiological alterations. Robust infection with P balayani -gt3ra was seen in rabbits when compared to experimental infection with P balayani gt3 and P balayani gt4. Consistent fecal viral shedding was observed more often than viremia, suggesting that fecal viral shedding could be the best predictor of persistent infection while studying chronic HEV infection [ 25 , 27 ].
Immunosuppression in rabbits was achieved by using cyclosporine A (Fig. 3 ). Rabbit strain P balayani -gt3ra demonstrated higher chronicity levels measured by the fecal viral shedding titers when compared to the zoonotic human derived P balayani gt3 and gt4. Liver fibrosis was evident in chronically infected rabbits [ 49 ].

Summary of immunosuppressed rabbit model with intravenous (IV) HEV inoculation. Chronic infection in rabbits with HEV gt3 leads to the development of fibrotic liver lesions. Cyclosporine A (CsA), rabbit (ra)
P balayani -gt3ra was found to replicate primarily in the intestine of rabbits and further disseminates to extrahepatic tissues. Interestingly, P balayani -gt3ra antigen was detected in the urine, kidney, cerebrospinal fluid but no such results were demonstrated in the rabbits infected with non-rabbit specific strains of P balayani gt3 and gt4 [ 49 ]. Surprisingly, higher numbers of single nucleotide variants (SNVs) were seen in the absence of immunosuppressants in rabbits infected with P balayani -gt3ra [ 49 ], highlighting the importance of the immune system in the development of quasispecies.
Recapitulation of human treatment using ribavirin for 3 months cleared HEV in cyclosporine treated rabbits. The Hecolin vaccine only provided partial protection when rabbits were already treated with immunosuppressive drugs. However, full protection was seen in rabbits against zoonotic P balayani gt3 and gt4 when vaccination was given before the start of the immunosuppressive treatment [ 49 ]. In addition, 94 differentially expressed genes (DEGs) and 10 hub genes (interacts with multiple genes and play an essential role in gene regulation and biological processes) have been found in rabbits with chronic HEV infection. The interferon signaling pathway and immune-related pathways were the primary areas of enrichment for DEGs in samples with chronic HEV infection. Most importantly, it was shown that Hub genes, such as MX1, OAS2, and IFI44, were involved in the pathophysiology of long-term HEV infection [ 82 ].
There are some disadvantages, even though immunocompromised rabbits challenged with P balayani -3ra are a very good model for simulating chronic HEV infection as seen in SOT patients. P balayani gt3 has been linked to rabbits yet attempts to infect rabbits experimentally with human strains of P balayani gt3 were not successful in these studies [ 76 , 83 , 84 ]. In contrast, another study demonstrated successful experimental infection in rabbits with human strains P balayani gt3 and gt4 [ 85 ]. This finding suggests that P balayani -3ra and swine or human P balayani -gt3 may differ in several biological aspects depending upon the host and virus species utilized for the studies. Furthermore, a larger animal is better suited to simulate the pathophysiology of a human disease than a smaller one because of the similarity in complexity between their organ structures. Additionally, there are few mechanistic rabbit investigations, especially those involving genetics, because there are few knockout or transgenic animals [ 80 ]. The rabbit is an excellent animal model, but despite its many benefits, it has not been employed extensively in HEV research yet. Compared to other smaller species like mice or guinea pigs, rabbits are more expensive to use due to the expense of the animal itself, larger and unique housing requirements (cage), and more difficult to maintain as they can be more prone to handling injuries than other species [ 80 ].
The murine model has become more popular in the last decade for HEV pathogenesis studies due to the advances in murine genetics and the abundance of tools and reagents available to study viral replication in mice. Lack of thymus and T lymphocytes in nude mice results in the absence of adaptive immune responses [ 86 ]. Furthermore, both functioning T and B cells are absent in severe combined immunodeficiency disease (SCID) mice [ 87 ]. Nude and SCID mouse innovations have led to preclinical and translational HEV research using these models in the last few decades.
P balayani gt4 was used to experimentally infect BALB/c nude mice, simulating human chronic HEV infection [ 50 ]. Increased levels of liver enzymes and HEV-specific antibodies in the blood suggested that the inoculated mice were undergoing more severe disease than contact mice. Liver histopathology revealed necrotic lesions with inflammation, which are similar to chronic HEV infection in human patients [ 50 ].
In 2016, experimental infection of zoonotic P balayani gt3 infection in human liver chimeric mice (mouse liver is partially populated with human cells) [ 88 ] demonstrated virus replication within 2 weeks. Virus derived from feces or liver were replication competent in comparison to plasma derived virus samples [ 51 ]. HEV RNA was consistently present in 100% of chimeric mouse livers from week 2 to week 14, and mouse passaged HEV was found to propagate for up to 100 days in vitro [ 51 ].
In 2020, BALB/c mice were used as a model for chronic P balayani gt4 HEV infection [ 52 ]. The mice were infected with a rhesus macaque-adapted gt4 chronically-mutated HEV strain [ 52 ]. HEV replication was efficient, and viral titers were persistently increased. HEV RNA was detected in various extrahepatic tissues, and HEV antigens were observed. The mice also showed enlarged portal tracts and proliferative fibrosis, and muted immune responses (reduced IFNα and IFNβ expression levels) [ 52 ].
In 2022, humanized uPA+/+-SCID mice were inoculated with the BeSW67HEV4-2008 viral strain [ 53 ]. The BeSW67HEV4-2008 strain, according to phylogenetic studies, is of the P balayani -4b subtype, whereas other strains that infect mice are of the P balayani -4 h subtype [ 53 ]. Interestingly, onset of infection and higher titers were observed in the mice inoculated with the mouse-passaged virus than the pig derived virus [ 53 ]. The virus’s adaptation to the human environment due to the close interaction of mice and humans may have improved infection of the mouse passaged virus.
Despite the promising results from the mouse models infected with HEV, there remain several drawbacks to these models that require further refinement. Lack of adaptive immune response in the humanized liver mice, skew towards a Th2 immune response in BALB/c mice and the predominant Th1 immune response in the C57BL/6 mice makes it difficult to understand the role of the adaptive immune system during HEV infections (Fig. 4 ) [ 92 ]. Intrasplenic and intraperitoneal inoculation used in the mouse models do not recapitulate the natural fecal oral transmission seen in humans. Interestingly, oral inoculum used in the human liver chimeric mice failed to establish infection [ 93 ]. Furthermore, the innate immunity in nude mice and the remnant natural killer (NK) cells in SCID mice limits the options for studying the host and viral interactions [ 94 ]. It has been further reported that there is a direct correlation between the age of nude mice with a drop in T cells [ 95 ]. Hence, even though immunosuppressed mice produce fibrosis recapitulating chronic HEV scenarios, multiple host related factors cannot be studied in mice.

Summary of immunosuppressed mouse model with oral, intravenous (IV), intrasplenic (IS) and intraperitoneal (IP) HEV inoculation. HEV gt4 infection in Balb/c mice were more prominent to develop chronic HEV infection demonstrating necrotic and fibrotic liver lesions
Rats are a natural host of Rocahepevirus ratti and have the potential to be an ideal candidate for the study of zoonotic rat HEV strains recently shown to spillover into humans. Immunosuppression of rats has been demonstrated with a combination of drugs that has been commonly used in human transplant patients. Drug combinations of prednisolone, tacrolimus, and mycophenolate mofetil have been successfully used to develop the immunosuppressed rat model demonstrating the phenotype seen in chronic hepatitis E patients via the inoculation of a rat specific strains; CCY and SRN (Fig. 5 ) [ 54 ].

Summary of immunosuppressive rat model with intravenous (IV) HEV inoculation. Chronically infected rats demonstrated the enhanced ALT liver enzyme level. Resolution of chronic infection was seen after the decrease in the immunosuppressive drugs which is the routine treatment regime in humans
Pathological lesions in the liver with some alteration in the liver enzymes such as ALT were observed in infected high dose immunosuppressed rats when compared to the low dose immunosuppressed and overall immunocompetent rats [ 54 ]. In addition, lack of viremia and restoration of immune responses were clear after stoppage of immunosuppressive drugs in rats mimicking the scenario seen in chronic HEV human patients [ 54 ]. Intraperitoneal ribavirin treatment further reduced viral suppression demonstrating the efficiency of the rat model to recapitulate human patient HEV infection [ 54 ].
In general, rats have become a popular choice for studying human disease because of their similarity with human genes involved in immunity, metabolic detoxification, chemosensation, and disease-linked human genes [ 93 ]. Being larger in size than mice make handling, sampling, and performing procedures easier. Recent developments in the availability of rat genomic tools have made it easier to manipulate the rat genome producing specific gene knockouts and knock-ins [ 94 ] allowing for more in depth understanding of disease factors at the genetic level. Although rat models have several advantages mimicking chronic disease, rats have a different coagulation system than humans leading to delayed wound healing [ 95 ]. In addition, immunosuppressed rats were shown to be not susceptible to the human derived P. balayani gt4 strains [ 54 ] highlighting its disadvantage in mimicking pathology using human circulating strains.
Mongolian gerbils
Mongolian gerbils are a newly reemerging animal model to study HEV induced acute [ 96 ] and neurological infection [ 97 ] associated with P. balayani genotypes. Of all available animal models, gerbils are the second smallest in body size after mice and thus easy to handle and study in significant numbers. Immunosuppression in gerbils was achieved by the surgical implantation of a tacrolimus pellet in the neck [ 55 ]. Mongolian gerbils were inoculated with the P. balayani gt3 strain (derived from macaques) (Fig. 6 ).

Summary of immunosuppressed gerbil model with intravenous (IV) HEV inoculation. CD68 + macrophage was absent in the liver of chronically infected gerbils. Extrahepatic distribution of HEV was seen in the chronically infected gerbils
Elevation of ALT in the blood, persistent viremia in concomitance with fecal viral shedding was reported in gerbils treated with tacrolimus [ 55 ]. Weak serological responses were reported for the immunosuppressed group when compared to immunocompetent gerbils. Interestingly, the immunological response in the liver was associated with the presence of CD68 + macrophages at microscopic foci demonstrating some apoptosis in the immunocompetent group. CD68 + macrophages were absent in the tacrolimus treated and infected group suggesting immunological damage in the liver of immunocompetent individuals during HEV infection [ 55 ]. Interestingly, the gerbil model has demonstrated experimental infection with a gerbil adapted gt1 strain. Pregnant gerbils infected with the gt1 strains developed robust, acute HEV infection and induced maternal mortality [ 98 ]. In addition, transmission of the virus to the offspring was noted [ 98 ]. These findings are very insightful and demonstrates the importance of pregnant gerbil models to understand the mechanism behind pregnancy mortality associated with HEV infection and to provide a mechanistic view of HEV crossing the blood placental barrier. In addition, tacrolimus prolonged HEV gt1 infection in gerbils [ 98 ], highlighting the scenario seen in humans where immunosuppressive drugs are known to lengthen the duration of infection leading to chronicity. Thus, the HEV gt1 infection gerbil model could be an interesting model to investigate the in-depth HEV immunopathogenesis, genotype associated pregnancy mortality, testing vaccines and antivirals against HEV.
As for model drawbacks, the implantation of the immunosuppressed drugs does not mimic the drug dosage routine in humans. The implantation of tacrolimus allows for continuous effects of the drug in body homeostasis [ 55 ]. One can argue that the small size of the gerbil, its potential to mimic active and neurological infection makes it a good animal model for acute infection. However, anatomical differences in the organ size, structure and functions when compared to humans and the availability of reagents make it a less popular immunosuppressed model for HEV.
Conclusions
The importance of chronically infected animal models in understanding HEV pathophysiology is of utmost importance in biomedical research. Advancements in the biomedical field have led to the development of immunosuppressed animal models either by the utilization of immunosuppressant drugs or by gene editing. To help the reader to understand the importance of utilizing a particular chronic HEV model, several potential questions that could be answered by the utilization of a model are listed below:
Potential questions answered by a chronic animal model recapitulating HEV infection | |
|---|---|
1. Does chronicity depend on HEV genotypes? | |
2. Are chronic liver lesions mediated by virus replication or the result of the host immune reaction? | |
3. Can pregnancy trigger HEV chronicity? | |
4. What are the roles of immune privileged sites during chronic HEV infection? | |
5. What is the role of HEV quasispecies in the maintenance of chronicity? | |
6. Can human derived HEV be infectious in an animal model recapitulating the chronic fecal viral shedding and viremia seen in humans? | |
7. Can higher antibody response and elevated liver enzymes be mimicked in an animal model? | |
8. Does reducing immunosuppression decrease HEV viral titers in an animal model? |
The above listed animal models such as cynomolgus monkeys, pigs, rabbits, mice, rats and gerbils have answered very important aspects of HEV pathophysiology during chronic HEV infection. Even though immunosuppressed cynomolgus monkeys, pigs, and rats recapitulate the chronic fecal viral shedding and viremia, they do not develop liver fibrosis as can be seen in chronic HEV model such as rabbits, mice, and gerbils. This suggests that even in animals recapitulating higher anatomical structure and physiological functions found in larger vertebrates more closely resembling humans, recapitulating all of the clinical manifestations of infectious disease is rare often necessitating multiple animals to answer some questions.
Existing literature suggests that the most chronic HEV cases are affiliated with zoonotic Paslahepevirus balayani gt3 and gt4 infections [ 3 ]. This suggests that the chronicity is related to certain genotypes of HEV. Recent advancement in understanding HEV have demonstrated the unique ability of HEV to cross the blood brain barrier (BBB) [ 99 ] and blood testis barrier (BTB) which are immune privileged sites in the body [ 100 ]. In addition, infectious HEV presence in the sperm head [ 101 ] demands the need to understand the ability of HEV to be transmitted sexually between partners. A recent study reported infectious HEV particles in semen of nine chronically infected men [ 102 ]. The viral load in semen was 100-fold higher when compared to the serum in five of the infected men [ 102 ]. Interestingly, few studies reported no evidence of the ability of HEV to transmit sexually between humans [ 103 , 104 ]. These studies highlighted HEV infection based on seroconversion but the demonstration of higher HEV titers in the ejaculate of chronically infected men delineates the need to understand the importance of HEV concentration, duration of shedding in ejaculate and ability to transmit the virus during active shedding of HEV in semen. Hence, the interplay between HEV and the immunosuppressed host needs to be further investigated by utilizing the immunosuppressed animal models.
With recent advancements in scientific technology, future studies in HEV need to be directed in specific areas: (a) the exact cellular receptor(s) that HEV recognizes and allows cellular entry. (b) the mechanisms by which HEV produces liver injury and cirrhosis in chronic HEV patients. (c) quasispecies formation in the central nervous system and its role in the devastating neurological effects in chronic HEV patients. (d) the ability of HEV to cross the BTB and the investigation of quasispecies and their role in the reinfection in chronic HEV patients. (e) the transmission route of HEV other than the fecal-oral, blood transfusions, and mother-to-child transmission. Chronic HEV infection in the immunosuppressed patient needs to be emphasized to understand the underlying role of viral factors leading to worse disease outcomes.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
Alanine Aminotransferase
Aspartate Aminotransferase
Blood Brain Barrier
Blood Testis Barrier
Differentially Expressed Genes
Gamma Glutamyl Transferase
Hepatitis E Virus
Intravenous
Open Reading Frame
Severe Combined Immunodeficiency Disease
Single Nucleotide Variants
Solid Organ Transplantation
Grange ZL, Goldstein T, Johnson CK, Anthony S, Gilardi K, Daszak P et al. Ranking the risk of animal-to-human spillover for newly discovered viruses. Proceedings of the National Academy of Sciences. 2021;118(15):e2002324118.
Lhomme S, Marion O, Abravanel F, Izopet J, Kamar N. Clinical manifestations, Pathogenesis and treatment of Hepatitis E Virus infections. J Clin Med. 2020;9(2).
Kamar N, Selves J, Mansuy JM, Ouezzani L, Péron JM, Guitard J, et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med. 2008;358(8):811–7.
Article CAS PubMed Google Scholar
Ollier L, Tieulie N, Sanderson F, Heudier P, Giordanengo V, Fuzibet JG, et al. Chronic hepatitis after hepatitis E virus infection in a patient with non-hodgkin lymphoma taking rituximab. Ann Intern Med. 2009;150(6):430–1.
Article PubMed Google Scholar
Dalton HR, Bendall RP, Keane FE, Tedder RS, Ijaz S. Persistent carriage of hepatitis E virus in patients with HIV infection. N Engl J Med. 2009;361(10):1025–7.
Thakur V, Ratho RK, Kumar S, Saxena SK, Bora I, Thakur P. Viral Hepatitis E and chronicity: a growing Public Health concern. Front Microbiol. 2020;11:577339.
Article PubMed PubMed Central Google Scholar
Thongprayoon C, Kaewput W, Pattharanitima P, Cheungpasitporn W. Progress and recent advances in solid organ transplantation. J Clin Med. 2022;11(8).
Tam AW, Smith MM, Guerra ME, Huang CC, Bradley DW, Fry KE, et al. Hepatitis E virus (HEV): molecular cloning and sequencing of the full-length viral genome. Virology. 1991;185(1):120–31.
Sridhar S, Yip CCY, Wu S, Cai J, Zhang AJ, Leung KH, et al. Rat Hepatitis E Virus as cause of Persistent Hepatitis after Liver Transplant. Emerg Infect Dis. 2018;24(12):2241–50.
Article CAS PubMed PubMed Central Google Scholar
Sridhar S, Yip CC, Wu S, Chew NF, Leung KH, Chan JF, et al. Transmission of Rat Hepatitis E virus infection to humans in Hong Kong: a clinical and epidemiological analysis. Hepatology (Baltimore MD). 2021;73(1):10–22.
Haagsma EB, van den Berg AP, Porte RJ, Benne CA, Vennema H, Reimerink JH, et al. Chronic hepatitis E virus infection in liver transplant recipients. Liver transplantation: official publication of the American Association for the study of Liver diseases. Int Liver Transplantation Soc. 2008;14(4):547–53.
Article Google Scholar
Lee GH, Tan BH, Teo EC, Lim SG, Dan YY, Wee A, et al. Chronic infection with Camelid Hepatitis E Virus in a liver transplant recipient who regularly consumes Camel meat and milk. Gastroenterology. 2016;150(2):355–e73.
Shirazi R, Pozzi P, Gozlan Y, Wax M, Lustig Y, Linial M et al. Identification of Hepatitis E virus genotypes 3 and 7 in Israel. Public Health Concern? Viruses. 2021;13(11).
Marion O, Lhomme S, Nayrac M, Dubois M, Pucelle M, Requena M, et al. Hepatitis E virus replication in human intestinal cells. Gut. 2020;69(5):901–10.
Kamar N, Dalton HR, Abravanel F, Izopet J. Hepatitis E virus infection. Clin Microbiol Rev. 2014;27(1):116–38.
Gérolami R, Moal V, Colson P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N Engl J Med. 2008;358(8):859–60.
Kamar N, Rostaing L, Legrand-Abravanel F, Izopet J. How should Hepatitis E virus infection be defined in organ-transplant recipients? Am J Transplantation: Official J Am Soc Transplantation Am Soc Transpl Surg. 2013;13(7):1935–6.
Article CAS Google Scholar
Kamar N, Garrouste C, Haagsma EB, Garrigue V, Pischke S, Chauvet C, et al. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology. 2011;140(5):1481–9.
Legrand-Abravanel F, Kamar N, Sandres-Saune K, Garrouste C, Dubois M, Mansuy JM, et al. Characteristics of autochthonous hepatitis E virus infection in solid-organ transplant recipients in France. J Infect Dis. 2010;202(6):835–44.
Pischke S, Stiefel P, Franz B, Bremer B, Suneetha PV, Heim A, et al. Chronic hepatitis e in heart transplant recipients. Am J Transplantation: Official J Am Soc Transplantation Am Soc Transpl Surg. 2012;12(11):3128–33.
Haagsma EB, Niesters HG, van den Berg AP, Riezebos-Brilman A, Porte RJ, Vennema H, et al. Prevalence of hepatitis E virus infection in liver transplant recipients. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver. Transplantation Soc. 2009;15(10):1225–8.
Google Scholar
Charre C, Ramière C, Dumortier J, Abravanel F, Lhomme S, Gincul R, et al. Chronic genotype 3 Hepatitis E in pregnant woman receiving infliximab and azathioprine. Emerg Infect Dis. 2018;24(5):941–3.
Mallet V, Le Mener S, Roque-Afonso AM, Tsatsaris V, Mamzer MF. Chronic hepatitis E infection cured by pregnancy. J Clin Virology: Official Publication Pan Am Soc Clin Virol. 2013;58(4):745–7.
Marion O, Abravanel F, Conan L, Dubucs C, Danjoux M, Izopet J, et al. Hepatitis E virus infection in a pregnant liver transplant recipient leading to chronic infection. Transplantation Direct. 2024;10(6):e1634.
Ambrosioni J, Mamin A, Hadengue A, Bernimoulin M, Samii K, Landelle C, et al. Long-term hepatitis E viral load kinetics in an immunocompromised patient treated with Ribavirin. Clin Microbiol Infection: Official Publication Eur Soc Clin Microbiol Infect Dis. 2014;20(10):O718–20.
Abravanel F, Lhomme S, Rostaing L, Kamar N, Izopet J. Protracted fecal shedding of HEV during Ribavirin therapy predicts treatment relapse. Clin Infect Diseases: Official Publication Infect Dis Soc Am. 2015;60(1):96–9.
Marion O, Lhomme S, Del Bello A, Abravanel F, Esposito L, Hébral AL, et al. Monitoring hepatitis E virus fecal shedding to optimize Ribavirin treatment duration in chronically infected transplant patients. J Hepatol. 2019;70(1):206–9.
Marion O, Capelli N, Lhomme S, Dubois M, Pucelle M, Abravanel F, et al. Hepatitis E virus genotype 3 and capsid protein in the blood and urine of immunocompromised patients. J Infect. 2019;78(3):232–40.
Zhang H, Rao H, Wang Y, Wang J, Kong X, Ji Y, et al. Evaluation of an antigen assay for diagnosing acute and chronic hepatitis E genotype 4 infection. J Gastroenterol Hepatol. 2019;34(2):458–65.
Kamar N, Mansuy JM, Cointault O, Selves J, Abravanel F, Danjoux M, et al. Hepatitis E virus-related cirrhosis in kidney- and kidney-pancreas-transplant recipients. Am J Transplantation: Official J Am Soc Transplantation Am Soc Transpl Surg. 2008;8(8):1744–8.
Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Reviews Gastroenterol Hepatol. 2021;18(3):151–66.
Pischke S, Suneetha PV, Baechlein C, Barg-Hock H, Heim A, Kamar N, et al. Hepatitis E virus infection as a cause of graft hepatitis in liver transplant recipients. Liver transplantation: official publication of the American Association for the study of Liver diseases. Int Liver Transplantation Soc. 2010;16(1):74–82.
Kamar N, Marion O, Abravanel F, Izopet J, Dalton HR. Extrahepatic manifestations of hepatitis E virus. Liver International: Official J Int Association Study Liver. 2016;36(4):467–72.
Damiris K, Aghaie Meybodi M, Niazi M, Pyrsopoulos N. Hepatitis E in immunocompromised individuals. World J Hepatol. 2022;14(3):482–94.
Alexandrova R, Tsachev I, Kirov P, Abudalleh A, Hristov H, Zhivkova T, et al. Hepatitis E Virus (HEV) infection among immunocompromised individuals: a brief narrative review. Infect drug Resist. 2024;17:1021–40.
Colson P, Payraudeau E, Leonnet C, De Montigny S, Villeneuve L, Motte A, et al. Severe thrombocytopenia associated with acute hepatitis E virus infection. J Clin Microbiol. 2008;46(7):2450–2.
Fourquet E, Mansuy JM, Bureau C, Recher C, Vinel JP, Izopet J et al. Severe thrombocytopenia associated with acute autochthonous hepatitis E. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology. 2010;48(1):73–4.
Ma Z, de Man RA, Kamar N, Pan Q. Chronic hepatitis E: advancing research and patient care. J Hepatol. 2022;77(4):1109–23.
Pischke S, Peron JM, von Wulffen M, von Felden J, Höner Zu Siederdissen C, Fournier S et al. Chronic Hepatitis E in Rheumatology and Internal Medicine patients: a Retrospective Multicenter European Cohort Study. Viruses. 2019;11(2).
Ghandili S, Lindhauer C, Pischke S, Zur Wiesch JS, Von Kroge PH, Polywka S, et al. Clinical features of hepatitis E infections in patients with hematologic disorders. Haematologica. 2022;107(12):2870–83.
Rivero-Juarez A, Lopez-Lopez P, Frias M, Rivero A. Hepatitis E infection in HIV-Infected patients. Front Microbiol. 2019;10:1425.
Cruz S, Campos C, Timóteo M, Tavares A, José Nascimento MS, Medeiros R, et al. Hepatitis E virus in hematopoietic stem cell transplant recipients: a systematic review. J Clin Virol. 2019;119:31–6.
Ian R, Fox T, Irish D, Bharucha T, Thorburn D, Mark H, et al. Hepatitis E Viraemia in Transplant recipients. Blood. 2016;128(22):3846.
Swartling L, Nordén R, Samuelsson E, Boriskina K, Valentini D, Westin J, et al. Hepatitis E virus is an infrequent but potentially serious infection in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2020;55(7):1255–63.
Michniacki TF, Choi SW, Peltier DC. Immune suppression in allogeneic hematopoietic stem cell transplantation. Handb Exp Pharmacol. 2022;272:209–43.
Gardinali NR, Guimarães JR, Melgaço JG, Kevorkian YB, Bottino FO, Vieira YR, et al. Cynomolgus monkeys are successfully and persistently infected with hepatitis E virus genotype 3 (HEV-3) after long-term immunosuppressive therapy. PLoS ONE. 2017;12(3):e0174070.
Cao D, Cao QM, Subramaniam S, Yugo DM, Heffron CL, Rogers AJ et al. Pig model mimicking chronic hepatitis E virus infection in immunocompromised patients to assess immune correlates during chronicity. Proceedings of the National Academy of Sciences. 2017;114(27):6914-23.
León-Janampa N, Caballero-Posadas I, Barc C, Darrouzain F, Moreau A, Guinoiseau T et al. A pig model of chronic hepatitis E displaying persistent viremia and a downregulation of innate immune responses in the liver. Hepatol Commun. 2023;7(11).
He Q, Zhang F, Shu J, Li S, Liang Z, Du M, et al. Immunocompromised rabbit model of chronic HEV reveals liver fibrosis and distinct efficacy of different vaccination strategies. Hepatology (Baltimore, Md; 2022.
Huang F, Zhang W, Gong G, Yuan C, Yan Y, Yang S, et al. Experimental infection of Balb/c nude mice with Hepatitis E virus. BMC Infect Dis. 2009;9:93.
Allweiss L, Gass S, Giersch K, Groth A, Kah J, Volz T, et al. Human liver chimeric mice as a new model of chronic hepatitis E virus infection and preclinical drug evaluation. J Hepatol. 2016;64(5):1033–40.
Li Y, Long F, Yang C, Hao X, Wu J, Situ J, et al. BALB/c mouse is a potential animal Model System for studying Acute and Chronic Genotype 4 Hepatitis E virus infection. Front Microbiol. 2020;11:1156.
Collignon L, Verhoye L, Hakze-Van der Honing R, Van der Poel WHM, Meuleman P. Study of Hepatitis E Virus-4 infection in human Liver-Chimeric, Immunodeficient, and Immunocompetent mice. Front Microbiol. 2022;13:819877.
Sridhar S, Wu S, Situ J, Shun EH, Li Z, Zhang AJ, et al. A small animal model of chronic hepatitis E infection using immunocompromised rats. JHEP Rep. 2022;4(10):100546.
Subramaniam S, Fares-Gusmao R, Sato S, Cullen JM, Takeda K, Farci P, et al. Distinct disease features of acute and persistent genotype 3 hepatitis E virus infection in immunocompetent and immunosuppressed Mongolian gerbils. PLoS Pathog. 2023;19(9):e1011664.
Krawczynski K, Meng XJ, Rybczynska J. Pathogenetic elements of hepatitis E and animal models of HEV infection. Virus Res. 2011;161(1):78–83.
Purcell RH, Engle RE, Govindarajan S, Herbert R, St Claire M, Elkins WR, et al. Pathobiology of hepatitis E: lessons learned from primate models. Emerg Microbes Infections. 2013;2(3):e9.
CAS Google Scholar
Tsarev SA, Tsareva TS, Emerson SU, Govindarajan S, Shapiro M, Gerin JL, et al. Successful passive and active immunization of cynomolgus monkeys against hepatitis E. Proc Natl Acad Sci U S A. 1994;91(21):10198–202.
Tsarev SA, Tsareva TS, Emerson SU, Yarbough PO, Legters LJ, Moskal T, et al. Infectivity titration of a prototype strain of hepatitis E virus in cynomolgus monkeys. J Med Virol. 1994;43(2):135–42.
Balayan MS, Andjaparidze AG, Savinskaya SS, Ketiladze ES, Braginsky DM, Savinov AP, et al. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology. 1983;20(1):23–31.
Kinugasa F, Nagatomi I, Ishikawa H, Nakanishi T, Maeda M, Hirose J, et al. Efficacy of oral treatment with tacrolimus in the renal transplant model in cynomolgus monkeys. J Pharmacol Sci. 2008;108(4):529–34.
Haustein SV, Kolterman AJ, Sundblad JJ, Fechner JH, Knechtle SJ. Nonhuman primate infections after organ transplantation. ILAR J. 2008;49(2):209–19.
Sykes M, Sachs DH. Transplanting organs from pigs to humans. Sci Immunol. 2019;4(41):eaau6298.
Pirenne J, Benedetti E, Gruessner A, Moon C, Hakim N, Fryer JP, et al. Combined transplantation of small and large bowel. FK506 versus cyclosporine A in a porcine model. Transplantation. 1996;61(12):1685–94.
Grant D, Duff J, Zhong R, Garcia B, Lipohar C, Keown P, et al. Successful intestinal transplantation in pigs treated with cyclosporine. Transplantation. 1988;45(2):279–84.
Dalton HR, Webb GW, Norton BC, Woolson KL, Hepatitis E, Virus. Time to change the textbooks. Digestive diseases. (Basel Switzerland). 2016;34(4):308–16.
Cao D, Cao QM, Subramaniam S, Yugo DM, Heffron CL, Rogers AJ, et al. Pig model mimicking chronic hepatitis E virus infection in immunocompromised patients to assess immune correlates during chronicity. Proc Natl Acad Sci U S A. 2017;114(27):6914–23.
Murali AR, Kotwal V, Chawla S. Chronic hepatitis E: a brief review. World J Hepatol. 2015;7(19):2194–201.
Singh A, Seth R, Gupta A, Shalimar, Nayak B, Acharya SK, et al. Chronic hepatitis E - an emerging disease in an immunocompromised host. Gastroenterol Rep. 2018;6(2):152–5.
Meng XJ, Hepatitis E. Virus: animal reservoirs and zoonotic risk. Vet Microbiol. 2010;140(3–4):256–65.
Jacobsen IA. Renal transplantation in the rabbit: a model for preservation studies. Lab Anim. 1978;12(2):63–70.
Kamaruzaman NA, Kardia E, Kamaldin NA, Latahir AZ, Yahaya BH. The rabbit as a model for studying lung disease and stem cell therapy. Biomed Res Int. 2013;2013:691830.
Jeklova E, Leva L, Jaglic Z, Faldyna M. Dexamethasone-induced immunosuppression: a rabbit model. Vet Immunol Immunopathol. 2008;122(3–4):231–40.
Graur D, Duret L, Gouy M. Phylogenetic position of the order Lagomorpha (rabbits, hares and allies). Nature. 1996;379(6563):333–5.
Ma H, Zheng L, Liu Y, Zhao C, Harrison TJ, Ma Y, et al. Experimental infection of rabbits with rabbit and genotypes 1 and 4 hepatitis E viruses. PLoS ONE. 2010;5(2):e9160–e.
Cheng X, Wang S, Dai X, Shi C, Wen Y, Zhu M, et al. Rabbit as a novel animal model for hepatitis E virus infection and vaccine evaluation. PLoS ONE. 2012;7(12):e51616–e.
Liu P, Bu QN, Wang L, Han J, Du RJ, Lei YX, et al. Transmission of hepatitis E virus from rabbits to cynomolgus macaques. Emerg Infect Dis. 2013;19(4):559–65.
Han J, Lei Y, Liu L, Liu P, Xia J, Zhang Y, et al. SPF rabbits infected with rabbit hepatitis E virus isolate experimentally showing the chronicity of hepatitis. PLoS ONE. 2014;9(6):e99861.
Xia J, Liu L, Wang L, Zhang Y, Zeng H, Liu P, et al. Experimental infection of pregnant rabbits with hepatitis E virus demonstrating high mortality and vertical transmission. J Viral Hepatitis. 2015;22(10):850–7.
Keir S, Page C. The rabbit as a model to study asthma and other lung diseases. Pulm Pharmacol Ther. 2008;21(5):721–30.
Karol MH. Animal models of occupational asthma. Eur Respir J. 1994;7(3):555–68.
Li M, Wang Y, Li K, Lan H, Zhou C. Characterization of highly expressed novel hub genes in hepatitis E virus chronicity in rabbits: a bioinformatics and experimental analysis. BMC Vet Res. 2022;18(1):239.
Smith DB, Simmonds P, Members Of The International Committee On The Taxonomy Of Viruses, Study G, Jameel S, Emerson SU, Harrison TJ, et al. Consensus proposals for classification of the family Hepeviridae. J Gen Virol. 2014;95(Pt 10):2223–32.
Zhang Y, Gong W, Song WT, Fu H, Wang L, Li M, et al. Different susceptibility and pathogenesis of rabbit genotype 3 hepatitis E virus (HEV-3) and human HEV-3 (JRC-HE3) in SPF rabbits. Vet Microbiol. 2017;207:1–6.
He Q, Zhang F, Shu J, Li S, Liang Z, Du M, et al. Immunocompromised rabbit model of chronic HEV reveals liver fibrosis and distinct efficacy of different vaccination strategies. Hepatology (Baltimore MD). 2022;76(3):788–802.
Flanagan SP. Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res. 1966;8(3):295–309.
Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301(5900):527–30.
Strom SC, Davila J, Grompe M. Chimeric mice with humanized liver: tools for the study of drug metabolism, excretion, and toxicity. Methods Mol Biol. 2010;640:491–509.
Sellers RS, Clifford CB, Treuting PM, Brayton C. Immunological variation between inbred laboratory mouse strains: points to consider in phenotyping genetically immunomodified mice. Vet Pathol. 2012;49(1):32–43.
Sayed IM, Verhoye L, Cocquerel L, Abravanel F, Foquet L, Montpellier C, et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut. 2017;66(5):920–9.
Budzynski W, Radzikowski C. Cytotoxic cells in immunodeficient athymic mice. Immunopharmacol Immunotoxicol. 1994;16(3):319–46.
Giovanella BC, Fogh J. The nude mouse in cancer research. Adv Cancer Res. 1985;44:69–120.
Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428(6982):493–521.
Parker CC, Chen H, Flagel SB, Geurts AM, Richards JB, Robinson TE, et al. Rats are the smart choice: Rationale for a renewed focus on rats in behavioral genetics. Neuropharmacology. 2014;76(0):250–8. Pt B(0.
Dorsett-Martin WA. Rat models of skin wound healing: a review. Wound Repair Regeneration. 2004;12(6):591–9.
Zhang W, Ami Y, Suzaki Y, Doan YH, Muramatsu M, Li TC. Mongolia Gerbils are broadly susceptible to Hepatitis E Virus. Viruses. 2022;14(6).
Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, et al. The evolutionary history of vertebrate RNA viruses. Nature. 2018;556(7700):197–202.
Liu T, He Q, Yang X, Li Y, Yuan D, Lu Q et al. An Immunocompetent Mongolian Gerbil Model for Hepatitis E Virus genotype 1 infection. Gastroenterology. 2024.
Tian D, Li W, Heffron CL, Wang B, Mahsoub HM, Sooryanarain H, et al. Hepatitis E virus infects brain microvascular endothelial cells, crosses the blood-brain barrier, and invades the central nervous system. Proc Natl Acad Sci U S A. 2022;119(24):e2201862119.
Horvatits T, Wißmann JE, Johne R, Groschup MH, Gadicherla AK, Schulze Zur Wiesch J, et al. Hepatitis E virus persists in the ejaculate of chronically infected men. J Hepatol. 2021;75(1):55–63.
Yadav KK, Boley PA, Laocharoensuk T, Khatiwada S, Lee CM, Bhandari M, et al. Infectious hepatitis E virus is associated with the mature sperm head. PLoS Pathog. 2024;20(5):e1012240.
Schemmerer M, Bock HH, Schattenberg JM, Huber S, Polywka S, Mader M, et al. Proof of infectivity of hepatitis E virus particles from the ejaculate of chronically infected patients. J Med Virol. 2024;96(6):e29735.
Schäfer G, Lübke R, Degen O, Mader M, Scheiter R, Wolski A, et al. Lack of evidence of acute HEV infections as a sexually transmitted disease: data from a German cohort of PrEP users. Brazilian J Infect Diseases: Official Publication Brazilian Soc Infect Dis. 2024;28(1):103720.
Chaix ML, Leturque N, Gabassi A, Charreau I, Minier M, Pialoux G et al. Prevalence and incidence of HEV among men using HIV pre-exposure prophylaxis: a sub-study of the ANRS IPERGAY trial. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology. 2023;160:105380.
Download references
Acknowledgements
Not applicable.
Additional salaries and research support were provided by state and federal funds appropriated to the Ohio Agricultural Research and Development Center, The Ohio State University, and from the research funds of National Institute of Allergy and Infectious Diseases (#R21AI151736).
Author information
Authors and affiliations.
Center for Food Animal Health, Department of Animal Sciences, The Ohio State University, 1680 Madison Ave, Wooster, OH, 44691, USA
Kush Kumar Yadav & Scott P. Kenney
Department of Veterinary Preventive Medicine, The Ohio State University, Columbus, 43210, USA
You can also search for this author in PubMed Google Scholar
Contributions
KKY and SK wrote the main manuscript text. KKY prepared the figures. KKY and SK revised the manuscript. SK provided the funding and oversight.
Corresponding author
Correspondence to Scott P. Kenney .
Ethics declarations
Ethics approval and consent to participate, consent for publication, authors information.
KKY has 6 years of experience working with HEV and has utilized the immunosuppressed pig animal model to understand the pathology of HEV ribavirin resistant mutant viruses. SPK has more than 14 years of experience working with HEV and has been awarded by the National Institute of Health (NIH) to study the pathogenesis of ribavirin resistant mutants in an immunosuppressed pig model. KKY and SPK have experiences with various animal models including but not limited to pigs, chickens, turkeys, rabbits, ferrets, mice, and hamsters.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ .
Reprints and permissions
About this article
Yadav, K.K., Kenney, S.P. Hepatitis E virus immunosuppressed animal models. BMC Infect Dis 24 , 965 (2024). https://doi.org/10.1186/s12879-024-09870-4
Download citation
Received : 31 May 2024
Accepted : 03 September 2024
Published : 12 September 2024
DOI : https://doi.org/10.1186/s12879-024-09870-4
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Immunosuppressed
Advertisement
- Find a journal
- Publish with us
- Track your research
Disclaimer: Early release articles are not considered as final versions. Any changes will be reflected in the online version in the month the article is officially released.
Volume 30, Number 10—October 2024
Evidence of Lineage 1 and 3 West Nile Virus in Person with Neuroinvasive Disease, Nebraska, United States, 2023
Suggested citation for this article
Suggested citation for this article : Davis E, Velez J, Hamik J, Fitzpatrick K, Haley J, Eschliman J, et al. Evidence of lineage 1 and 3 West Nile virus in person with neuroinvasive disease, Nebraska, United States, 2023. Emerg Infect Dis. 2024 Oct [ date cited ]. https://doi.org/10.3201/eid3010.240595
DOI: 10.3201/eid3010.240595
Table of Contents – Volume 30, Number 10—October 2024
| EID Search Options |
|---|
| – Search articles by author and/or keyword. |
| – Search articles by the topic country. |
| – Search articles by article type and issue. |
Metric Details
Article views: 139.
Data is collected weekly and does not include downloads and attachments. View data is from .
What is the Altmetric Attention Score?
The Altmetric Attention Score for a research output provides an indicator of the amount of attention that it has received. The score is derived from an automated algorithm, and represents a weighted count of the amount of attention Altmetric picked up for a research output.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
The PMC website is updating on October 15, 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Oxford University Press - PMC COVID-19 Collection
Emerging Challenges and Opportunities in Infectious Disease Epidemiology
Joseph a lewnard.
Division of Epidemiology and Biostatistics, School of Public Health, University of California, Berkeley, Berkeley, California
Arthur L Reingold
Much of the intellectual tradition of modern epidemiology stems from efforts to understand and combat chronic diseases persisting through the 20th century epidemiologic transition of countries such as the United States and United Kingdom. After decades of relative obscurity, infectious disease epidemiology has undergone an intellectual rebirth in recent years amid increasing recognition of the threat posed by both new and familiar pathogens. Here, we review the emerging coalescence of infectious disease epidemiology around a core set of study designs and statistical methods bearing little resemblance to the chronic disease epidemiology toolkit. We offer our outlook on challenges and opportunities facing the field, including the integration of novel molecular and digital information sources into disease surveillance, the assimilation of such data into models of pathogen spread, and the increasing contribution of models to public health practice. We next consider emerging paradigms in causal inference for infectious diseases, ranging from approaches to evaluating vaccines and antimicrobial therapies to the task of ascribing clinical syndromes to etiologic microorganisms, an age-old problem transformed by our increasing ability to characterize human-associated microbiota. These areas represent an increasingly important component of epidemiology training programs for future generations of researchers and practitioners.
The priority afforded to infectious diseases within epidemiologic research has been fluid over the past 200 years or longer. Despite the lasting prominence of early investigations into measles, cholera, plague, typhoid fever, malaria, and yellow fever ( 1 – 6 ), the intellectual tradition of modern epidemiology stems largely from studies of chronic diseases dating to the post-World War II era, when such conditions came to surpass infectious diseases in morbidity and mortality in high-income countries amid improvements in living conditions and the introduction of numerous antibiotics and vaccines. This epidemiologic transition co-occurred with a shift in focus for epidemiologic research (and training programs) toward the multifactorial etiology of chronic conditions ( 7 ). In parallel, early 20th-century work on the “dependent happenings” of communicable diseases ( 8 – 11 ) yielded to the development of today’s core biostatistical methods for chronic diseases, premised on the independence of outcomes among subjects ( 12 – 14 ).
Since the late 20th century, the emergence of human immunodeficiency virus and acquired immunodeficiency syndrome and other infections has renewed interest in infectious diseases and their health, economic, and security implications ( 15 ). Outbreaks of severe acute respiratory syndrome, pandemic influenza A H1N1, Ebola virus (EBOV), and Zika virus have prompted international responses, whereas influenza A H7N9 and H5N1, Lassa fever virus, Nipah virus, and Middle East respiratory syndrome coronavirus, among other agents, have been a source of regional concern. The incidence and endemic range of “neglected” infections, including dengue and cholera ( 16 , 17 ), have expanded, and antimicrobial resistance has threatened to derail the control of tuberculosis, typhoid, malaria, gonorrhea, yaws, and invasive bacterial infections ( 18 – 23 ). Although highly effective vaccines are available, measles and yellow fever have resurged on multiple continents, due to gaps in vaccine coverage ( 24 , 25 ), while short-lived vaccine-induced protection has facilitated unexpected resurgences in diseases once on the path to elimination, such as pertussis and mumps ( 26 , 27 ).
After decades of relative obscurity in the mid-20th century, infectious disease epidemiology has experienced an intellectual rebirth in response to disease emergence. Repopulation of this field by scientists trained not only in clinical medicine but ecology, demography, and quantitative sciences has led to the adoption of methods scarcely addressed in traditional public health training programs. Cluster-randomized trial designs, for example, have become commonplace for evaluating infectious disease interventions, quantifying indirect effects resulting from contagion ( 28 ). Classical models of ecological dynamics have been adapted to address the transmission and control of infectious agents, while our expanding ability to integrate such models with epidemiologic data through Bayesian statistics has enhanced their relevance to policymaking ( 29 – 31 ). Most recently, sequencing and phylogenetic analysis have afforded an unprecedented view into the population structure and dynamics of pathogens ( 32 , 33 ).
Here, we review developments in infectious disease epidemiology together with their implications for research and practice, and for the training of future epidemiologists. We first consider the role of epidemiologic surveillance in the context of pathogen emergence and the integration of surveillance data into quantitative studies of transmission. Next, we discuss challenges in causal inference, including the evaluation of public health interventions against emerging pathogens and the difficulties of attributing clinical syndromes to microbial agents.
SURVEILLANCE OF EMERGING INFECTIONS
Public health practice.
Surveillance justifiably has been seen as a core public health function, with its important role articulated by Langmuir in 1963 ( 34 ). In the United States and elsewhere, surveillance for diverse infectious (and noninfectious) diseases has typically relied on an essentially passive system of reporting by health-care providers or laboratories, often mandated by public health laws. Although such passive systems of disease reporting have proven invaluable, their limitations, such as incomplete, often inconsistent detection of cases and delayed detection of outbreaks, are well documented ( 35 ). As a result, active surveillance systems that do not depend on providers and laboratories to report have been developed and promoted.
The Centers for Disease Control and Prevention–funded Emerging Infections Program and its components (e.g., Foodnet, ABCs), initiated in 1994, is 1 notable example of such a system, as are international versions promoted through the Centers for Disease Control and Prevention’s Global Health Security initiative ( 36 ). Such systems, while expensive to develop and maintain, are of great value, especially when they collect biological specimens (e.g., isolates of bacterial and viral pathogens) for typing and support analytic epidemiologic studies (e.g., case-control studies of vaccine effectiveness ( 37 )). Nevertheless, these systems, too, may have limitations, particularly in their ability to detect in a timely fashion outbreaks caused by novel microbial agents, prompting interest in alternative methods. In this article, we consider proposed alternatives, highlighting their benefits and challenges.
Surveillance for disease emergence
Beyond efforts to quantify the incidence of infectious diseases of known etiology, disease surveillance has been advocated as a means of mitigating the threat posed by novel pathogens. Large-scale efforts to identify pathogens with the potential to spill over from animals to humans have received notable investment, for instance from the US Agency for International Development Emerging Pandemic Threats program ( 38 ). More recently, metagenomic sequencing has enabled the number of known viruses to be multiplied in such studies ( 39 ). The aim of those working on the Global Virome Project, launched in 2018, to characterize within 10 years all 1.6 million viruses thought to exist ( 40 ).
However, the pathway for translating data sets produced by such activities into actionable threat-reduction programs remains unclear. That only approximately 250 viruses are known to infect humans (and an even smaller handful to cause major epidemics) may constrain the value of large-scale virus discovery for identifying high-risk pathogens, as well as viral determinants of pathogenic potential ( 41 ). Spillover events causing recent epidemics—including H1N1 in Mexico, Middle East respiratory syndrome in Saudi Arabia, and EBOV in West Africa—have been poorly predicted by factors long believed to drive disease emergence ( 42 ). Accordingly, interest in expanding our catalogue of potential pathogens should be weighed against our persisting need to enhance the detection and control of outbreaks of known pathogens ( 43 ). For instance, 2 EBOV epidemics in the Democratic Republic of the Congo in 2018 took weeks to be identified, with dozens of suspected cases having already accumulated ( 44 , 45 ).
Serological studies have received increasing enthusiasm for monitoring emerging pathogens of significance to humans ( 41 , 46 ). The prevalence of antibodies indicating previous exposure may provide valuable information about the frequency of animal-human spillover events and the potential for person-to-person spread, overcoming reporting biases that favor detection of large outbreaks under traditional surveillance. Moreover, the low cost of multiplex assays makes integrated surveillance of multiple pathogens plausible. Although serosurveys have bolstered recent efforts to understand the geographic range and clinical spectrum of EBOV and Zika virus infections ( 47 , 48 ), the enhancement of dengue hemorrhagic fever risk by prior exposure ( 49 ), and the role of immunologic history in influenza susceptibility and vaccine response ( 50 ), there remain few examples of public health programs undertaking serological studies for routine surveillance, at least in civilian populations ( 51 ).
Emerging data and analytics
Outside of laboratory-based surveillance, the increasing availability of passively collected “Big Data” on the health and behaviors of individuals has prompted enthusiasm about enhancing disease surveillance through alternative data streams. Initiatives such as the ProMED-mail network and HealthMap ( 52 , 53 ) compile and disseminate news about outbreaks from media and other sources, aiming to trigger investigation by public health organizations. Data such as emergency department visits, medication sales, online search queries, and social media postings have also been suggested as real-time indicators of outbreak activity, although their integration into public health responses remains a subject of debate ( 54 – 56 ). The need to overcome reporting biases is a central challenge, because observations may be too nonspecific to distinguish between meaningful and spurious signals in settings with high technological capacity while also being insensitive to even high-risk events in resource-poor settings ( 57 , 58 ). Although nontraditional data sources have, in some applications, supported inferences about epidemic dynamics ( 59 ), limited information about cases from such sources remains a barrier. For instance, models fitted from news reports of recent measles and mumps outbreaks have yielded considerable underestimates of vaccine coverage ( 60 – 62 ), underscoring the importance of field investigations.
Forecasting the incidence of diseases has been a more successful application of these emerging data streams and data-analytic approaches. Although the acknowledged failure of Google Flu Trends—a prediction approach based on Internet search behavior—yielded important lessons about nonmechanistic forecasts, approaches based on machine learning and crowd-sourced human judgment have provided the most accurate within-season predictions of US influenza activity in recent comparisons ( 63 – 65 ). Given expanding interest in forecasting among researchers, funding agencies, and other stakeholders, there is a clear and compelling need to evaluate whether such forecasts can enhance the success and efficiency of public health response efforts.
UNDERSTANDING TRANSMISSION DYNAMICS
Model-data integration for emerging diseases.
Mathematical modeling as a means to understanding infectious disease spread dates to studies by Sir Ronald Ross ( 8 ). Although the use of models to connect data such as age of infection to transmission dynamics of endemic infections has longstanding precedent ( 66 , 67 ), assimilation of outbreak data for near-term assessments of control priorities is a comparatively recent phenomenon. Integration of modeling with the public health response to epidemics of bovine spongiform encephalopathy and foot-and-mouth disease in the United Kingdom and the severe acute respiratory syndrome epidemic ( 68 – 73 ) has led to expectations for near real-time modeling studies during major outbreaks. In recent experience, models of the spread of pandemic influenza A H1N1 ( 74 ), cholera ( 75 ), Middle East respiratory syndrome ( 76 ), EBOV ( 77 , 78 ), Chikungunya virus ( 79 ), Zika virus ( 80 , 81 ), yellow fever ( 82 ), and plague ( 83 ) have all been published within weeks of the respective outbreak notifications.
Although the circumstances of particular epidemics dictate what data may be available and pertinent, methods for fitting models to data have generally focused on exponential growth rates in cases ( 84 ) or the distribution of the serial interval ( 85 ). Methods based on the latter class of data offer the advantage of illustrating real-time changes in reproductive numbers ( 86 ); however, the requisite information from patient line lists is seldom available. Reliance instead on ecological data exposes models to numerous vulnerabilities, notably the inability to discern individual risk factors (and thus the population meaningfully at risk). These shortcomings may prevent models from predicting reductions in transmission before depletion of the susceptible population.
A challenge thus lies ahead in determining the role of models in outbreak response and the best practices for communicating modeling results. Although the ability of models to evaluate prophylactic strategies may be considered a benefit, recommendations to act against remote future risks have sometimes triggered resistance among stakeholders ( 87 ). During the West African EBOV epidemic, for example, attention to worst-case model-based projections prompted some to question the reliability of the models ( 88 ), reflecting an important discrepancy between public understanding of modeling as a forecasting tool and the intended uses of models for scenario-based comparisons ( 89 , 90 ). This use of modeling has been better understood in attempts to communicate the impact of interventions after the fact ( 91 ).
Microbial sequencing
The ease of sequencing pathogen genomes has afforded a new view into transmission during outbreaks. Use of sequence data to identify transmission clusters in the presence of unsure epidemiologic links dates to the early years of the human immunodeficiency virus and acquired immunodeficiency syndrome epidemic ( 92 ). In recent years, sequencing has aided efforts to track the sources of unexplained epidemics of cholera in Haiti ( 93 ) and EBOV in West Africa ( 94 ), and has shown increasing utility for reconstructing the geographic spread of pathogens ( 95 , 96 ). A particular advantage of phylogenetic analysis is the possibility of estimating unobserved epidemiologic quantities, such as the reporting fraction ( 97 ) and reproductive numbers for subcritical transmission ( 98 ), which remain difficult to assess from traditional case-notification data.
Beyond reconstructing the demographic history of pathogen lineages, recent years have seen progress toward joint analysis of epidemiologic and sequencing data ( 99 ). Such “phylodynamic” approaches have shown particular relevance for emerging infections, including distinguishing the role of repeated introductions and subsequent local transmission ( 100 – 103 ). Whereas most applications have been tailored to specific data sets and assumptions, the development of generalized methods for joint inference of epidemiologic and phylogenetic parameters remains a priority ( 104 ) to support real-time analysis.
EVALUATION OF INFECTIOUS DISEASE INTERVENTIONS
Efficacy evaluations in emergencies.
The ability to rapidly develop and deploy countermeasures to mitigate the threat posed by emerging infections has received increasing recognition as a component of public health preparedness. However, outbreaks are difficult environments in which to evaluate interventions. During the West African EBOV epidemic, the feasibility of a new paradigm for development and evaluation of interventions in emergencies was demonstrated by accelerated vaccine safety, immunogenicity, and efficacy studies( 105 ). The Coalition for Epidemic Preparedness Innovations was established in 2017, with an initial focus on vaccines against Nipah virus, Middle East respiratory syndrome coronavirus, and Lassa fever virus, in addition to adaptable vaccine platforms for novel threats ( 106 ).
Lessons learned in EBOV vaccine trials will have an influential bearing on evaluations during future emergencies. Despite efforts to accelerate evaluation of candidate vaccines, incidence had reached low levels by the time phase III efficacy trials were ready to begin, posing a threat to their statistical power: A planned trial in Liberia was canceled due to declining transmission ( 107 ), and no cases of disease occurred in a second trial in Sierra Leone ( 108 ), preventing efficacy assessments. In a stepped-wedge trial in Guinea, clusters of primary and secondary contacts of EBOV disease cases were randomly assigned to immediate or delayed vaccination; no cases were reported among vaccine recipients during the trial ( 109 ) or in subsequent field deployments of the vaccine, supporting a conclusion of near 100% vaccine efficacy.
Debates surrounding design of these trials highlight methodological questions requiring additional attention. In the Guinean trial, a “ring” vaccination scheme helped maximize power by enrolling contacts of known cases ( 110 ). However, the choice of individual- or cluster-level randomization within rings was debated. Because members of a vaccinated cluster are exposed to direct protection through vaccination and indirect protection due to reduced transmission within their clusters ( 28 ), cluster-randomized trials have weaker statistical power than individually randomized trials of the same size ( 111 ). Moreover, the direct effect measured in individually randomized studies may be a preferred, transportable efficacy measure ( 112 ). Uses of simulation helped in planning vaccine trials tailored to the real-world circumstances of the EBOV outbreak ( 113 ) and enabled trialists to compare alternative designs in terms of ethical mandates ( 114 ). Simulation-guided design further presents the opportunity for applying adaptive trial methods ( 115 ) in the context of infectious disease outbreaks, where dynamic trends in incidence may highlight the benefits of such approaches.
Observational designs
In addition to efficacy trials for new interventions, observational studies are needed to assess licensed interventions against evolving and re-emerging pathogens. Most commonly applied in evaluations of influenza vaccines, test-negative designs have become popular in routine ( 116 ) and exploratory ( 117 ) studies of vaccine effectiveness. By measuring vaccine effectiveness from the exposure odds ratio of vaccination among individuals seeking care who test positive or negative for a pathogen of interest, this design seeks to overcome associations of health-care seeking with vaccination status ( 118 ). However, it is uncertain whether health-care seeking and other sources of confounding are appropriately controlled for, and whether measures accurately capture vaccine direct effects ( 119 , 120 ). Uncertainty about the validity of estimates that routinely inform vaccine policy-making demonstrates the need for formal evaluations of such studies and strategies to reduce bias.
Time series analyses of public health surveillance data provide another approach to measuring the real-world impact of vaccination on disease incidence, with the advantage of identifying the overall effect of a vaccination program resulting from direct and indirect protection ( 28 ). Although the ecological nature of such designs permits the introduction of biases from changes in diagnostic practices or health-care seeking, such studies nonetheless have offered important insights where other approaches failed. Limited reductions in influenza-related deaths among elderly persons amid increases in influenza vaccine coverage during the 1990s provided an important indication that the “healthy vaccinee” effect accounted for astonishing and implausible protection against all-cause mortality among elderly influenza vaccine recipients in cohort and case-control studies ( 121 – 123 ). Newer methods continue to improve public health inferences obtained from time-series data. In a recent evaluation of invasive pneumococcal disease incidence, trends expected under continued use of 7-valent pneumococcal conjugate vaccine provided a counterfactual condition for measuring the impact of the switch to a 13-valent vaccine targeting emerging serotypes ( 124 ). Bayesian averaging of models encoding differing pre- and postvaccination trends and change points provides a generalized strategy for defining such counterfactual comparisons ( 125 ). Other signals of transmission intensity in surveillance data, such as age of infection and sub- or multiannual periodicity, may provide additional insights while reducing sensitivity to fluctuations in reporting effort ( 126 , 127 ).
The re-emergence of pathogens against which vaccines are widely deployed, such as varicella, pertussis, and mumps in the United States, poses additional challenges for conducting vaccine effectiveness studies. Situational factors may undermine researchers’ ability to establish the extent to which cases owe to primary or secondary vaccine failure in all or certain vaccine recipients, and whether emerging pathogen lineages are escaping vaccine-driven immune pressure. For instance, high compliance with vaccine schedules may limit variation in individuals’ vaccination status and exposures, necessitating large samples to detect factors influencing vaccine performance ( 128 ). Because re-emergence most likely reflects the expansion of 1 or several pathogen clades, limited pathogen diversity may hinder the application of conventional approaches to identifying microbial determinants of vaccine escape ( 129 , 130 ). Novel methods to distinguish null from vaccine-driven mutations in antigen-coding regions ( 102 , 131 ) may streamline efforts to identify vaccine escape, while mathematical modeling provides a basis for comparing candidate hypotheses with observations ( 26 , 27 ).
Vaccine safety
Because vaccine studies are typically powered for primary clinical endpoints, long-term observational studies are needed to monitor for rare vaccine-attributable adverse events. Such studies have been crucial to identifying safety concerns such as intussusception after rotavirus vaccination ( 132 ) and to refuting spurious links, such as autism onset after measles-mumps-rubella vaccination ( 133 ). However, unique challenges arise in vaccine safety studies; at the individual level, vaccination and adverse-event detection may be confounded due to health-care–seeking behavior, whereas at the population level, age-related confounding may occur when vaccine recommendations are based on the individual’s age.
Ecological designs taking advantage of natural experiments have proven useful in numerous studies of vaccine safety ( 134 , 135 ) but inconclusive for certain classes of rare events, including those that also result from the vaccine-targeted infection ( 136 , 137 ). Recent years have seen growing interest in case-only methods offering the ability to reduce or rule out individual-level sources of confounding. Case-crossover methods are common among such approaches ( 138 , 139 ) and resemble matched case-control studies by sampling “control” periods from the person-time contributed by case individuals before an adverse event. Self-controlled case-series methods similarly benefit from the use of cases as their own controls, following a cohort logic in estimating the relative incidence of adverse events after vaccination within specified risk periods ( 140 ); with adequate sample size, researchers may be able to use such analyses to eliminate or greatly reduce potential time- or age-related confounding.
Antimicrobial drugs
In response to the growing threat posed by antimicrobial resistance, the World Health Organization and national governments have prioritized bringing novel antimicrobial drugs to market ( 141 ). These plans will necessitate phase III trials in which the efficacy of new therapeutic agents is addressed and possibly phase IV studies, in which the optimal use of new and existing drugs, either singly or in combination, can be determined. Whereas patients traditionally have been enrolled in antimicrobial treatment studies on the basis of target bacterial species infections or clinical syndromes, it is uncertain within what strata such trials may yield transportable inferences. Rather than merely the infecting pathogen’s baseline resistance or susceptibility phenotype, strata may be defined by factors such as pathogen lineage, mutational barriers to resistance development ( 142 ), and presence of horizontally transferable resistance elements in cocolonizing agents or environmental sources ( 143 ). Stratification based on interpatient and even intrapatient tumor heterogeneity is an emerging feature of cancer therapy trials and may provide a template for such designs ( 144 ).
In addition to clinical endpoints, carriage of susceptible and resistant bacteria, including commensal agents not purposefully targeted by treatment, can inform the impact of treatment on resistance selection in targeted and bystander species ( 145 ). Whereas between-group differences in the absolute prevalence of colonization with resistant organisms ( 146 ) are tested for routinely, stratified measurements (see the reports of Shrag et al. ( 147 ) and Feikin et al. ( 148 ), for example) of the effect of treatment on acquisition and clearance of susceptible and resistant pathogens are more informative of the underlying biology ( 149 ) and may detect signals of selection masked by simpler between-group comparisons ( 150 , 151 ).
Studies are also needed to address optimal deployment of new and existing antimicrobial drugs in clinical practice. The tradeoff between maximizing a drug’s impact and minimizing resistance selection has led policymakers to ration certain new drugs as last-resort treatments. However, in recent experience, such decisions have ignited ethical debates ( 152 ). Coupling mathematical modeling with field-based studies has proven useful for understanding the effectiveness of antimicrobial use policies, as highlighted in recent evaluations of the risk of resistance under population-wide access of the tuberculosis drug bedaquiline ( 153 , 154 ) and antimicrobial cycling to limit resistance selection in hospital settings ( 155 , 156 ).
ETIOLOGIC UNDERSTANDING
Whereas certain efforts we have discussed have been made to identify novel microorganisms able to cause human infection ( 40 , 157 ), most epidemics caused by emerging pathogens have been recognized first by clusters of anomalous syndromes—such as cardiopulmonary syndrome caused by New World hantaviruses, severe acute respiratory syndrome caused by a coronavirus, and congenital abnormalities caused by Zika virus —before the role or even existence of the etiologic microorganism had been characterized. The problem of ascribing a clinical syndrome to an etiologic agent is among the oldest in epidemiology, dating at least to the 19th century, when Robert Koch laid out criteria for such inference (i.e., Koch’s, or more correctly, Henle-Koch’s, postulates ( 158 )). However, these postulates have long been recognized as inadequate, particularly for illnesses caused by viruses, and thus of largely historical interest ( 159 ); for instance, the notion that a pathogen should be absent from healthy individuals is incompatible with the prominence of carriage and asymptomatic infection in the natural history of numerous pathogens. A growing appreciation of the complexity of the human microbiome, and the likelihood that intricate mixtures of microorganisms at diverse body sites may be either the cause or consequence of both detrimental and beneficial physiological states, has further highlighted the difficulty of linking a given health outcome to infection by a single microorganism.
The now-recognized critically important role of persistent infection and the inflammation it can produce in diverse cancers and possibly other chronic diseases has further diminished the relevance of Koch’s postulates and the age-old distinction between infectious and chronic diseases, as illustrated in the well-known example of human papillomavirus causing cervical, anal, and oral cancers ( 160 ). The best causal understanding of such relationships has come from randomized trials demonstrating that infection-preventing interventions are efficacious against downstream chronic illness, such as peptic ulcers due to Helicobacter pylori and chronic wheeze due to respiratory syncytial virus ( 161 , 162 ). Natural experiments following the same intuition have provided additional evidence of such relationships, such as measles-induced immunosuppression ( 163 ), malnutrition and stunting due to enteric infection ( 164 ), and complex or chronic otitis media due to tissue damage from acute early-life disease ( 165 ). Such relationships have proven difficult to probe in the absence of a randomized or natural experiment, because of the likelihood that confounding factors influence individuals’ risk for initial infection as well as chronic sequelae. New paradigms for ascribing an etiologic role to microorganisms and resulting host responses are clearly needed and may prove important in efforts to quantify the health impacts of infectious disease interventions.
The recognition in the 1970s and 1980s that infectious diseases were not, in fact, disappearing as important causes of morbidity and mortality in human populations has been followed by renewed interest in these conditions, especially in the emergence or re-emergence of diverse infectious diseases and in the role of infection in various chronic diseases. At the same time, advances in epidemiologic and statistical methods, together with the growing availability of data from diverse sources, have provided new tools and approaches for studying infectious diseases. The infectious disease epidemiologists of the future will need a solid grounding in the biology of infection and the host immune response, as well as training in the increasingly sophisticated approaches to causal inference; the manipulation and analysis of large-scale data sets, including pathogen genome sequences; and mathematical modeling, together with the behavioral and social determinants of health. Integration of these elements into epidemiology training programs (e.g., through coursework in Bayesian statistics and phylogenetics) represents an increasingly important consideration for academic departments.
ACKNOWLEDGMENTS
Authors’ affiliations: Division of Epidemiology and Biostatistics, School of Public Health, University of California, Berkeley, Berkeley, California (Joseph A. Lewnard, Arthur L. Reingold).
The authors received no specific funding for this article.
Conflict of interest: none declared.
Abbreviation
| EBOV | Ebola virus |
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Open access
- Published: 16 September 2024
Crossing epigenetic frontiers: the intersection of novel histone modifications and diseases
- Weiyi Yao 1 ,
- Xinting Hu 1 , 2 &
- Xin Wang ORCID: orcid.org/0000-0001-8051-1481 1 , 2 , 3
Signal Transduction and Targeted Therapy volume 9 , Article number: 232 ( 2024 ) Cite this article
1 Altmetric
Metrics details
- Cancer therapy
- Drug delivery
- Epigenetics analysis
Histone post-translational modifications (HPTMs), as one of the core mechanisms of epigenetic regulation, are garnering increasing attention due to their close association with the onset and progression of diseases and their potential as targeted therapeutic agents. Advances in high-throughput molecular tools and the abundance of bioinformatics data have led to the discovery of novel HPTMs which similarly affect gene expression, metabolism, and chromatin structure. Furthermore, a growing body of research has demonstrated that novel histone modifications also play crucial roles in the development and progression of various diseases, including various cancers, cardiovascular diseases, infectious diseases, psychiatric disorders, and reproductive system diseases. This review defines nine novel histone modifications: lactylation, citrullination, crotonylation, succinylation, SUMOylation, propionylation, butyrylation, 2-hydroxyisobutyrylation, and 2-hydroxybutyrylation. It comprehensively introduces the modification processes of these nine novel HPTMs, their roles in transcription, replication, DNA repair and recombination, metabolism, and chromatin structure, as well as their involvement in promoting the occurrence and development of various diseases and their clinical applications as therapeutic targets and potential biomarkers. Moreover, this review provides a detailed overview of novel HPTM inhibitors targeting various targets and their emerging strategies in the treatment of multiple diseases while offering insights into their future development prospects and challenges. Additionally, we briefly introduce novel epigenetic research techniques and their applications in the field of novel HPTM research.
Similar content being viewed by others

Histone lysine methyltransferases in biology and disease

The language of chromatin modification in human cancers

Histone post-translational modifications — cause and consequence of genome function
Introduction.
Histones, as the fundamental building blocks of chromatin, have their functions and characteristics regulated through an intricate and complex array of HPTMs. 1 These modifications work in concert to determine the spatial structure of chromatin and profoundly influence the expression and interpretation of genetic information. 2 The diverse changes brought about by HPTMs offer an additional layer of control over gene expression, enabling cells to respond flexibly to various external stimuli. This adaptability ensures the normal functioning of vital processes while also allowing for rapid reactions to environmental changes. 3 , 4 , 5 Proteins undergo various HPTMs that are crucial for chromatin regulation and function, affecting gene expression, chromatin structure, and cellular responses to stimuli. While acetylation (ac) and methylation (ma) are well-studied HPTMs, there has been a recent realization that the functions of many HPTMs remain largely unknown. Newly discovered HPTMs include a range of lysine ac such as propionylation (pr), butyrylation (bu), crotonylation (cr), hydroxyisobutyrylation (hib), succinylation (succ), and ma. 6 , 7 , 8 , 9 Many of these ac sites coincide with known ac sites, prompting questions regarding the functional similarities between them. 10 HPTMs are subject to influence by external environmental factors and metabolic status, and they possess the ability to significantly affect the initiation and progression of a range of conditions, including inflammation, 11 cancers, 12 cardiovascular diseases, 13 kidney diseases, 14 metabolic disorders 15 and neuropsychiatric diseases. 16
HPTMs serve as a pivotal link between cellular metabolism and the control of epigenetic mechanisms, emerging as a focal point of study within this domain. 17 In the realm of cancer treatment, notably, inhibitors that target HPTMs, including ma and ac, have shown efficacy. For example, demethylating agents and histone deacetylase inhibitors can effectively treat acute myeloid leukemia and T-cell lymphoma. 18 Furthermore, targeting epigenetic regulatory factors represents an effective strategy for reversing drug resistance. 19 These advantages have prompted researchers to shift their focus towards the emerging field of HPTMs studies in recent years. With breakthroughs in proteomics research using high-resolution mass spectrometry (HRMS), research has identified nine novel HPTMs, 20 including lactylation (la), 21 citrullination (cit), cr, 22 succ, 9 pr, 6 bu, 6 SUMOylation, hib 8 and 2-hydroxybutyrylation (bhb). 23 Recent studies have elucidated the novel mechanistic roles, associations with cancers, and potential therapeutic applications of these HPTMs, revealing substantial practical implications.
The developmental trajectory of Hptms
The concept of epigenetics, initially introduced by Conrad Hal Waddington in the year 1942 as a mechanism elucidating how the genotype begets the phenotype, has experienced substantial development. From Waddington’s designation of epigenetics as the process through which the genotype engenders the phenotype, 24 to Nanney’s 25 focus on the regulatory systems governing gene expression, and further to Riggs, 26 Holliday, 27 , 28 Martienssen, and Russo emphasizing heritable gene function changes that cannot be explained by alterations in the DNA sequence. Bird 29 construed epigenetics as the adaptive alterations of chromosomal structures, while Greally and Lappalainen posited that it involves gene regulators that endow cells with the capacity to memorialize past events. Lastly, Nicoglou highlighted the stability exerted by intracellular factors on the potentialities of the genome. These definitions collectively mirror the richness and complexity inherent in the domain of epigenetics. 30 Waddington’s concept of the “epigenetic landscape,” introduced in 1957, emphasizes that cellular differentiation could be regulated by alterations in the epigenetic landscape rather than by changes in the genes themselves. 24 , 31 , 32 Subsequent advancements in epigenetics have resulted in breakthroughs across various aspects: In 1964, the first description of histone modifications closely linked to the regulation of RNA synthesis; 33 the chromatin nucleosome organization model proposed in 1974, which detailed the basic unit of chromatin; 34 the discovery of DNA modifications in 1975, particularly 5-methylcytosine, demonstrating its relevance to gene regulation; 28 and in 1976, Sanger’s identification of circular RNA, along with the first long non-coding RNA (H19) recognized in 1990, blazed new trails for epigenetic regulatory research. 35 , 36 In the year 1994, the unveiling of the initial microRNA, lin-4, illuminated the process through which miRNAs orchestrate the regulation of gene expression. This is achieved by their complementary association with specific target mRNAs. 36 Additionally, the initial discovery in 1996 of histone acetyltransferases (HATs) and histone deacetylases (HDACs) provided insights into the role of protein acetylation in epigenetics. 37 , 38 The year 1997 marked a significant advancement in the realm of molecular biology, with X-ray crystallography elucidating the nucleosome core particle’s structure within chromatin. This revelation provided a profound insight into the organizational framework of DNA and the intricacies of its regulatory systems. 39 Since the turn of the millennium, the field of epigenetics has continued to evolve rapidly. The discovery of SUV39H1 marked the beginning of histone lysine methyltransferase research, with the first histone lysine demethylase LSD1 following closely behind in 2004. 32 , 40 In 2006, the FDA approved the first batch of epigenetic drugs for cancer treatment, and in 2012, the first reports of cancer-related histone gene mutations emerged. 41 , 42 In 2015, the NIH Roadmap Epigenomics Mapping Consortium released 111 reference human epigenomes. 43 Over the past five years, a multitude of epigenetic drugs targeting DNA methyltransferases, HDACs, HMTs, and BET proteins have been tested in clinical trials, investigating their efficacy in treating various diseases, both as monotherapies and in combination therapies. 30 These achievements reflect the multifaceted nature of epigenetics as a concept across different disciplinary contexts, revealing the historical evolution and rich definitions within this field.
Before the early 1990s, it was widely held that histones—compact basic proteins that, in conjunction with DNA, constitute the chromatin framework in the nucleus—simply functioned as structural support for DNA, playing no active role in the regulation of genes. 44 However, subsequent research has shown that histones play crucial roles in gene expression regulation, DNA damage repair, DNA replication, and recombination. Histones are pivotal mediators in the epigenetic regulatory landscape, influencing inheritable chromatin configurations that transcend the genetic script of DNA. Such a role is indispensable for cellular differentiation, where histones undergo an array of covalent modifications. These alterations span phosphorylation, ubiquitination, acetylation, methylation, along with emergent types of histone modifications such as la, cit, cr, succ, pr, bu, SUMOylation, hib, and bhb. The histone code hypothesis posits that these modifications, occurring singly or in concert on one or several histone tails, operate in a sequential or combinatorial manner, creating a ‘histone code’. This code is interpreted by specific proteins, which then initiate diverse downstream biological outcomes. The resulting histone codes may manifest as transient signals or as more enduring entities, with the latter embodying the true heritable epigenetic code. 45
Histone modifications, as an integral component of epigenetics, were first described in 1964 when histone ac was discovered, playing a crucial role in local chromatin relaxation. This process of ac, by neutralizing the positive charges on lysine residues, diminishes the interaction strength between histones and DNA. 46 However, it was not until between 1996 and 1998 that, with the development of molecular cloning techniques and protein purification methods, scientists successfully identified and characterized eight different HATs containing acetyltransferase domains. This significant advancement not only enriched our understanding of the regulatory functions of histones but also unveiled the pivotal role of ac in gene expression. Regarding histone ma, its functional role was recognized as early as 1962, but a deeper investigation into its mechanisms became possible only with the identification of HMTs. These HMTs catalyze the ma of histones at specific residues, thereby regulating gene expression and chromatin structure. From 1993 to 2005, scientists gradually identified a range of HMTs responsible for various ma reactions, broadening our comprehension of the complexity and precision of histone functions. As we ventured into the new millennium, a succession of pivotal discoveries concerning novel histone modifications has been made. These have not only enriched our comprehension of cellular biology but have also unveiled new vistas for therapeutic intervention. To begin with, in 2003, Shiio and Eisenman conducted pioneering research on the SUMOylation of histone H4 at lysine 12, marking the commencement of explorations into the mechanisms of histone modification. 47 Subsequently, in 2007, Chen and colleagues discovered the ubiquitination and proline isomerization modifications of histones, further enriching our understanding of the diversity of histone functions. By 2011, scientists had for the first time identified the succ of histones in mammals, offering a fresh perspective on the regulation of histone modifications. 48 However, in 2014, Dai’s team, utilizing advanced mass spectrometry techniques and chemical biology methods, identified a novel histone modification known as bu, thereby pioneering a new field in histone modification research. 6 Subsequently, in 2016, Xie and colleagues identified bhb as a HPTM, further expanding our understanding of the diversity of histone modifications. 23 By 2017, Tan and others, through a systematic analysis, discovered a new HPTM, cr, highlighting the importance of specific histone modifications in the regulation of gene expression. 7 Finally, in 2019, a study revealed that an accumulation of lactate could trigger histone la, affecting gene transcription and thereby elucidating the critical role of HPTMs in diseases such as cancer and inflammation. 21 Fundamentally, this continuum of discoveries underscores the cumulative and forward-moving nature of scientific inquiry. Each stride is scaffolded upon pre-existing knowledge, methodically disclosing the intricate and consequential role played by histone modifications in the orchestration of gene expression and cellular operations. These studies not only provide new theoretical perspectives in biology but also pave the way for future biomedical research ( Fig. 1 ) .

A succinct historical overview of the development of epigenetics and HPTMs. a Key discoveries in histone modifications and chromatin biology from the 1960s to 2020s. b Chronological overview oflandmark events in the field of epigenetics, including conceptual developments and practical applications in medicine
Hptms: epigenetics’ key components
Structure of histones.
Epigenetic regulation primarily occurs through four key mechanisms: ma of DNA, modifications of histone post-translationally (often referred to as HPTMs), modulation of the chromatin architecture, and the regulation by noncoding RNAs. 49 , 50 Histones, essential components of epigenetic machinations, fulfill two critical nuclear roles: DNA compaction and gene expression regulation. These small, basic proteins are characterized by a globular domain at the C-terminus and a tail at the N-terminus. Within the eukaryotic cell nucleus, they associate with DNA, constructing nucleosomes—the fundamental building blocks of chromatin. Approximately 146 base pairs of DNA coil around a core histone octamer, comprised of two each of histones H2A and H2B, and a tetramer of histones H3 and H4. Additionally, the linker histone H1 fortifies chromatin structure by anchoring both to the nucleosome and the inter-nucleosomal DNA. 51 Histones are adorned with a variety of HPTMs, primarily occurring on their N-terminal tails. 52 Histone tails, characterized by their inherent disordered structure and extensive post-translational modifications, are central to transcriptional regulation. 53 These modifications, which include both activation through positive regulation and suppression via negative mechanisms, endow histones with a dynamic regulatory capacity. Predominantly positively charged due to rich lysine and arginine residues, with about two-thirds of these charges located in the tails, histones modulate interactions with DNA and among themselves, thereby intricately influencing gene activation and repression. In essence, the modifications of histone tails are crucial for the nuanced balance of gene expression control. 54
In the preceding decade and a half, scientific exploration has evolved markedly. Where initial inquiries were once centered on the HPTMs of histone tails, contemporary studies have shed light on the functional consequences of modifications occurring within the globular domains of histones. The lateral surfaces of the histone octamer, in direct contact with DNA, pose greater challenges in terms of accessibility compared to the more exposed histone tails. Despite this, nucleosomes are dynamic structures; their DNA intermittently unwinds from and rebinds to the lateral surface, consequently providing chromatin-modifying enzymes the opportunity to access and alter these nucleosomal regions. 55 Numerous HPTMs discerned within the globular domain appear to be situated on the lateral surface or at the junctures between the histones forming the octamer. This observation intimates that alongside the extensively researched histone tail modifications, those within the globular domains are equally integral to the dynamic behavior of nucleosomes. These modifications influence the interactions between DNA and histones, as well as the overall structure and function of chromatin, exemplified by modifications on the lateral surface of the histone octamer, 56 HPTMs surrounding the symmetry axis, 57 and modifications at the octamer interfaces. 58
Furthermore, the codex of histone modifications is characterized by its dynamism and complexity. 50 For instance, histones can undergo a multitude of modifications on different amino acid residues. Mass spectrometry has unveiled intricate patterns of modifications on histones H2A, H2B, H3, and H4, identifying them as having 13, 12, 21, and 14 potential modification sites, respectively. 59 These modifications encompass ma, ac, phosphorylation, and ubiquitination, among others. Each site may undergo modification or remain unmodified, thereby amplifying the potentialities and intricacies of these modifications. Notably, lysine residues can not only be methylated or acetylated but, in the case of ma, can exhibit variability with mono-, di-, or trime. Given these combinations and variations, the overall pattern of histone modifications displays a tremendous diversity, constituting a complex regulatory network. Additionally, the code of histone modifications can sometimes change transiently with the cellular milieu, reflecting shifts in the cell’s physiological state and surrounding signals. 29 Furthermore, research has unveiled that specific “active” histone modification patterns, including H3K4ma2/ma3, H3K79ma2, and ac of H3 and H4, exhibit temporal stability in the promoter regions of genes, persisting even in cells halted in mitosis. This suggests the potential inheritability of such modification patterns. 60 , 61 The concept of an inheritable histone code is embraced as an epigenetic code, denoting the transmission of this code from cell to cell across disparate tissues within multicellular entities. Consequently, this influences the expression tableau across the organism’s entirety. 45
Enzymes in Hptms
HPTMs are of paramount importance in epigenetic discourse, as they modulate chromatin architecture and gene activity by appending chemical groups to amino acid residues on histone tails. The dynamism of these modifications involves a variety of specialized enzymes, primarily categorized into three groups: “readers,” “writers,” and “erasers“. 62 “Reader” enzymes recognize and bind to specific chemical modifications, such as ma or ac, on histones, thereby recruiting other proteins or protein complexes to the chromatin and subsequently influencing gene activity. This recognition mechanism is facilitated through specialized domains, enabling precise regulation of transcriptional activity by the “reader” enzymes. “Writer” enzymes are responsible for adding new chemical modifications, such as methyl, acetyl, or phosphate groups, to histones. Enzymes like SETD2, KDM5B, and EP300 play key roles in cellular function and development by catalyzing specific lysine or serine residues and regulating gene expression through these modifications. In contrast, “eraser” enzymes are tasked with removing existing histone modifications. “Erasers” such as demethylases and deacetylases, like HDAC3, affect gene expression and modulate cellular functions by removing modifications, like acetyl groups, from histones. 63 , 64 The activity of these enzymes is crucial for maintaining the dynamic state of the chromatin and for adaptive modulation of gene expression patterns. 49
Within the realm of epigenetic regulation, HPTMs involve a myriad of specific enzymes tasked with catalyzing, recognizing, or removing modifications. P300/CBP, as a ubiquitously present acyltransferase, readily facilitates the production of histone pr and bu. 65 , 66 The GCN5 and MYST families are specifically dedicated to the targeted inscription of pr. 67 Within the context of histone ma and succ, the corresponding malonyl-CoA and succinyl-CoA regulate the levels of these modifications under the catalytic influence of acyltransferases. SIRT5 possesses the capacity for demalonylation, while SIRT2 demonstrates similar functionality in yeast. 68 In the context of bhb, the enzyme P300 adeptly appends β-hydroxybutyryl moieties, whereas SIRT3 is adept at reversing this biochemical process. 69 Additionally, class I and III HDACs harbor the capacity to excise β-hydroxybutyrate moieties. The regulation of histone glutarylation is managed by GCN5 and SIRT7, with glutaryl-CoA being a determinant in the modulation of glutarylation’s prevalence and localization. 58 Furthermore, the recognition and catalysis of cr involve the YEATS and DPF domains, as well as CDYL reader structures, with P300 considered to be the sole known octanoyltransferase. 70 In conclusion, the deacylation of crotonylated histone peptides and proteins is predominantly executed by members of the SIRT family, including SIRT1, SIRT2, and SIRT3. Conversely, the overexpression of HDACs can diminish the concentration of cr marks. 71 , 72 The activity of these enzymes is integral to the nuanced regulation of gene expression and profoundly influences cellular metabolism and development, occupying a central position in the context of cellular physiology and pathology.
The role of dysregulated novel HPTMs in disease pathophysiology
Histone lactylation.
Lactate is a common byproduct of glycolysis, predominantly converted from pyruvate through the action of lactate dehydrogenase (LDH). 73 Despite traditionally being considered a metabolic waste product, lactate exhibits anomalous glycolytic behavior in tumor cells, characterized by overproduction even in the presence of ample oxygen—a phenomenon first identified by Otto Warburg, known as the “Warburg effect“. 74 In recent years, lactate’s role in regulating various intracellular and extracellular dynamic processes has been increasingly recognized, encompassing gene expression, metabolic dynamics, the tumor microenvironment (TME), as well as the activation or inhibition of immune cells. 75 , 76
Zhang and colleagues’ research has uncovered histone lysine la as a novel epigenetic modification, which directly originates from exogenous or endogenous lactate. 21 By employing carbon-13 labeled lactate and glucose, combined with mass spectrometry analysis, the study confirmed that lactate can be converted into lactyl groups on histones. This discovery not only validates the existence of histone la but also provides a new perspective on the intracellular functions of lactate. Further investigation revealed that the dynamics of la and ac differ, with these modifications being regulated differently by glucose metabolism. 77 The research team also explored the relationship between lactate production and the levels of histone la, finding that glycolytic inhibitors, which reduce lactate generation, decrease the levels of histone la, whereas mitochondrial inhibitors or hypoxic conditions that increase lactate production led to an increase in histone la. Moreover, the study highlighted that various histone lysine ac, such as bu, pr, and hib, are associated with changes in the rate of glycolysis. 7 , 69 Recent studies indicate that controlling the concentration of acetyl coenzyme A (the substrate for acetyltransferases) can effectively regulate the levels of histone ac. 78 Overall, the research conducted by Zhang and colleagues illuminates the significant role of histone la in cellular metabolism and epigenetic regulation, particularly under hypoxic conditions, where an increase in lactate independently of the acetyl coenzyme A regulatory pathway directly influences the elevation of histone la levels. These findings pave the way for future research into how lactate can affect cellular functions through epigenetic mechanisms.
Lactate is a known energy source for cancer cells, which shuttle lactate to adjacent cancer cells, the surrounding stroma, and vascular endothelial cells, thereby inducing metabolic reprogramming. 79 Lactate not only contributes to the promotion of tumor-associated inflammation but also serves as a signaling molecule that can stimulate angiogenesis within the tumor milieu. However, the non-metabolic effects of lactate at high concentrations remain unclear. 80 In 2019, studies showed lactate accumulation triggers histone la, influencing gene transcription, and suggesting its significant role in conditions like cancer and inflammation. 21 Additionally, many HPTMs are not enzymatically regulated but are merely chemical by-products. Additionally, metabolic intermediates serve not only as sources of chemical substances but also regulate gene expression by influencing HPTMs. Evidence of “writers” and “erasers” confirms that their regulation is at least partially enzyme-mediated, thereby making HPTMs both targetable and directly exploitable. 81 , 82 Recent research connects histone la to cancer progression, emphasizing lysine la’s importance in altering cancer cell metabolism. 83 Disruption of histone la unbalances gene transcription, leading to cancer and various diseases. Recent research connects histone la to cancer progression, emphasizing lysine la’s importance in altering cancer cell metabolism. 83 Disruption of histone la unbalances gene transcription, leading to cancer and various diseases. Moreover, existing studies indicate a strong link between histone la and cancer prognosis.
Histone citrullination
Histone citrullination is a HPTM catalyzed by the peptidylarginine deiminase (PAD) family, a process contingent upon elevated calcium concentrations. Under pathological conditions, PAD enzymes have the ability to citrullinate various structural proteins. 84 Studies suggest that histone cit is implicated in autoimmune conditions, exemplified by rheumatoid arthritis (RA), wherein the presence of anti-citrullinated protein antibodies is a distinct marker for the disease. They can be utilized for early detection, reflect disease outcomes, and serve as valuable diagnostic and prognostic tools for RA. 85 , 86 Considering the involvement of PADs in both normal physiological functions and disease processes, research and application of PAD inhibitors have been pursued. Numerous PAD inhibitors are utilized for the treatment of PAD-associated diseases in the skin, joints, colon, and immune system. 87 , 88 , 89 , 90
PADs are regarded as transcription-regulating proteins that influence gene expression, the precise biological functions of citrullination remain unclear. 91 Citrullinated histones account for about 10% of all histone molecules in HL-60 granulocytes, underscoring the significance of this HPTMs in numerous nucleus-associated processes. 92 Cit plays a vital role in embryonic development; studies have demonstrated that the use of a specific PAD1 inhibitor significantly reduces cit of H4R3 and H3R2/8/17 in embryonic cells, coinciding with a developmental arrest at the four-cell stage. 93 Moreover, in a zebrafish tissue injury model, PAD2-mediated H4 cit is essential for effective regeneration, posited as a potential intermediary between early calcium signaling and subsequent wound healing. 94 The linker histone H1.0, when citrullinated, accumulates in aging cells and is involved in heterochromatinization and the aging process. 95 PAD2 modulates the expression of genes related to lactation through histone cit. 96 , 97 Another function of citrullinated histones is to modulate the formation of neutrophil extracellular traps (NETs). 98 , 99 In innate immunity, neutrophils are the first responders to bacterial infection, combating various pathogens by forming so-called NETs. 98 The enzyme PAD4-mediated cit of histones plays a pivotal role in the genesis of NETs. Neutrophils deficient in PAD4 fail to produce NETs upon exposure to chemotactic stimuli or bacterial incubation, highlighting the essentiality of PAD4 in the NET-dependent antibacterial response. 100 Research has also discovered that inhibiting PAD2 can reduce the formation of NETs and the production of inflammatory cytokines in sepsis, suggesting that PAD2 may play an important role in regulating both NET formation and inflammatory responses. 101 In cases of COVID-19, an increase in the quantity of NET remnants has been observed in patients’ serum, indicating that healthy neutrophils, upon exposure to serum samples from COVID-19 patients, undergo NETosis more frequently. 102 Histone cit contributes significantly to embryonic development, reproductive functions, chromatin expression, dissolution, pluripotency, and the formation of NETs.
Histone cit by PAD enzymes is intricately linked to cancer progression, impacting tumor development, gene regulation, cell differentiation and cell death, and plays a key role in chromatin activity modulation. PAD inhibitors have demonstrated immense potential in the field of cancer therapy, with PAD4 inhibitors in particular being applied to prevent tumor metastasis and associated thrombosis in cancer patients. 103 Histone cit by PAD is increasingly recognized as a diagnostic marker and treatment target in cancer research. Investigations have revealed that in a spectrum of malignancies—among them non-small cell lung cancer (NSCLC), gastric cancer, hepatitis B virus-associated hepatocellular carcinoma (HCC), and various malignant hematological disorders—PAD-mediated cit of histones is notably elevated. 104
NETs are observed in a variety of human cancer types, recognized as contributors to cancer progression. 105 , 106 PAD4, an enzyme abundantly found in various cancers and neutrophils, plays a critical role in the formation of NETs. 95 Additionally, the development of NETs driven by histone cit is closely associated with tumor proliferation and spread, involving processes such as ECM reorganization, surgical stress response, hypoxic conditions, alterations in fatty acids and cellular interactions through physical binding. 104 Corroborating this perspective, Demers et al. utilized PAD4-deficient mice, which intrinsically exhibit impaired neutrophil chromatin decondensation and diminished NETs generation capabilities. Their findings insinuated that when neutrophils initiate NETosis, it inadvertently promotes tumor proliferation. Thus, the study suggested that tumors, or the environment they exist in, trigger neutrophils to undergo NETosis, leading to a build-up of NETs within the tumor itself, which in turn promotes tumor proliferation. 107 In summary, histone cit contributes to cancer progression and dissemination through NET production.
Histone crotonylation
Histone cr was first identified by Tan and colleagues through the analysis of MS data. Utilizing the PTMap software to pinpoint post-translational modification sites, they uncovered 28 potential butyrylated histone markers. 7 , 108 Tan and colleagues have discovered that novel histone modifications mark TSS of active genes with specificity for cr, predominantly located at active promoters, which are critical to the regulation of gene expression. This finding underscores the significant role of histone cr in modulating chromatin structure and function. 7 , 109 Histone cr, much like ac, invariably takes place on lysine residues and is dynamically governed by the enzymatic actions of crotonyltransferases and decrotonylases. 110 Cr is differentiated from ac within the histone modification landscape by its distinct structural features, including a four-carbon planar chain and an alkenyl C = C double bond, which confer upon it unique functional properties. 70 In the mechanistic dance of cr, crotonyltransferases orchestrate the donation of crotonyl groups from crotonyl-CoA to specific amino acid residues on histones, while decrotonylases play their part in excising these crotonyl moieties, ensuring a dynamic equilibrium. HATs also play a regulatory role in the cr of histones, while a class of HDAC1, 2, 3, 8 operate as decrotonylases. Furthermore, studies have found that the concentration of crotonyl-CoA is a limiting factor in histone cr. 111 , 112 Cr is a highly dynamic modification, capable of either activating or repressing transcription. Cr exerts a more significant impact on the regulation of cell cycle and metabolism compared to ac, with its influence being dependent on particular genes or environmental conditions. In both human somatic cells and mouse male germ cells, cr is observed at the promoter regions of actively transcribed genes or at enhancers, where it plays a pivotal role in the modulation of gene transcription. 7 , 111 Histone cr is a critical element in orchestrating a multitude of biological pathways, including the response to acute renal damage, the maturation of sperm cells, the preservation of chromosomal end structures, the dormancy of the human immunodeficiency virus, and the advancement of oncological diseases. 14 , 113 , 114 , 115 , 116 , 117
Histone succinylation
The phenomenon of histone succ first emerged in scientific literature with its identification in mammals in 2011. 48 Histone succ was first identified in the activity of homoserine succinyltransferase and subsequently confirmed as a natural and novel post-translational modification through methods such as Western blot analysis, isotopic labeling, tandem MS/MS, and co-elution experiments using high-performance liquid chromatography (HPLC). 118 , 119 This entity has been detected across diverse biological systems, ranging from the bacterium Escherichia coli, unicellular yeast organisms, the protozoan Toxoplasma gondii, to the cultured human cervical cancer cells known as HeLa, and extending to the hepatic tissues of mice. 9 , 120 Discoveries across various cell types indicate a high degree of evolutionary conservation among species. Many of the identified succ sites overlap with sites of other HPTMs, such as ac, ma, and hib, suggesting intricate interactions between histone modifications. The identification of lysine succ has also led to the discovery of other similar modifications, such as pr and bu. 121 Succ can exert a direct influence on the organization of chromatin by chemically altering histones or by modulating interactions within the nucleus. This process can induce spatial conformational changes within the chromatin structure that are more substantial than those caused by ma or ac. 9 , 69 The presence of succinyl groups on lysine residues can attenuate the interaction between DNA and histones, thereby destabilizing nucleosomes and chromosomal integrity. This weakening of associations permits DNA to disengage more readily from its protein constraints, enhancing the accessibility of transcription factors to DNA sequences. The consequence is an upregulation in the transcriptional activity of genes. 122
Numerous studies support the notion that enzymes can catalyze the succ of histones. For instance, p300 can induce succ on synthetic histone tail peptides in vitro. 121 , 123 , 124 Atsushi Yokoyama and colleagues have provided compelling evidence that the succ of nuclear histones is an enzymatically driven process. 125 Moreover, studies spearheaded by Wang et al. have illuminated that the enzyme lysine acetyltransferase 2 A (KAT2A), also recognized as GCN5, possesses a preferential binding affinity for succinyl-CoA over acetyl-CoA. 126 The α-ketoglutarate dehydrogenase complex, comprising three constituent components, facilitates the production of succinyl-CoA within the nucleus, thus supplying the required substrate for KAT2A to catalyze histone succ. Contrastingly, a segment of research posits that enzymatic action might not be pivotal in the succ of histones. Research by Simithy et al. has found that the succ of histones H3 and H4 is primarily mediated by non-enzymatic actions under conditions involving a variety of HATs and non-enzymatic circumstances. 127 , 128 Elevating the concentration of succinyl-CoA can increase the abundance of nuclear succ. Acidic acyl modifications, including ma, succ, and glutarylation, are more amenable to non-enzymatic catalysis in the absence of enzymes.
Histone succ orchestrated by KAT2A might be a key player in disease pathogenesis, as seen in the emergence of human pancreatic ductal adenocarcinoma (PDAC) and in the context of Hepatitis B virus infection. KAT2A’s significant influence on gene regulation and cell proliferation is exemplified by the observation that, within PDAC samples, the succ at histone H3 lysine 79 augments the levels of 14-3-3ζ and β-catenin. This modification is correlated with the modulation of cellular glycolytic pathways and the enhancement of cellular migration and invasiveness. 129 Research shows histone succ affects various biological processes, including protease function and gene control, influencing diseases like cancer, cardiac, hepatic disorders and neurodegeneration. 130 Recent studies increasingly show histone succ’s key role in tumor growth and progression, suggesting new paths for cancer treatment.
Histone SUMOylation
Histone SUMOylation, the bonding of SUMO proteins to histones discovered by Shiio and Eisenman in 2003. They investigated the SUMOylation of histone H4 at lysine 12 and discussed the potential role of this modification in gene repression. 47
Although histone ma, ac, and ubiquitination have been extensively studied, research into histone SUMOylation has been challenging due to its extremely low abundance in cells and the lack of specific antibodies. 47 , 131 Histone SUMOylation occurs across a variety of organisms, including yeast, protozoa, and plants, and is involved in the regulation of transcription, centromere assembly, chromatin structure modulation, and double-strand break repair, among other functions. A specific lysine residue, K12, in histone H4 emerges as a major recurrent site of SUMOylation, which is typically associated with the suppression of gene transcription. Biophysical studies have suggested that H4K12succ is incompatible with the compact chromatin structures linked to transcriptional silencing. Further biochemical investigations have revealed that H4K12succ enhances the activity of the specific histone demethylase LSD1 within nucleosomes for H3K4ma2. 132 Evidence has shown that H4K12succ directly inhibits transcription mediated by RNA Polymerase II on chromatin templates and engages in direct negative crosstalk with p300-mediated histone ac and Set1/COMPASS-mediated histone ma, modifications typically associated with active gene transcription. 133
Histone propionylation and histone butyrylation
In 2007, Chen et al. were the first to discover bu and pr of histones. 6 These HPTMs display a considerable level of evolutionary conservation across eukaryotic species, as evidenced by their presence in yeast cells, murine hepatic tissue, and the U937 human leukemia cell line. 65 , 134 , 135 Propionyl-CoA and butyryl-CoA act as substrates, donating the propionyl and butyryl moieties respectively, which are indispensable for the ac reactions leading to protein pr and bu. 69 Histone lysine pr and bu are chemically similar to ac, with the addition of extra carbon atoms, suggesting that they may serve as analogs to ma. 6 , 136 These HPTMs are proposed to be catalyzed in vitro by HATs, although evidence of their occurrence in vivo remains unclear. 137 The activity of enzymes that modify histones, such as HATs, is regulated by the concentration of intracellular metabolites, such as acetyl coenzyme A, which act as cofactors for the enzymes, thereby linking the chromatin state to cellular metabolism. 138 , 139 Propionyl CoA and butyryl CoA, related to acylation, serve as intermediates in fatty acid and amino acid metabolism. Studies identifying and mapping H3K14pr in vivo indicate its enrichment at active TSS and promoters within the mouse liver, correlating it with transcriptional activity across various metabolic states. 135 While H3K14pr attracts specific binding partners, the role of H3K14bu remains less understood. It may prevent the recruitment of certain complexes, or possibly attract other binding proteins not identified in this study. Fluctuations in global H3K14pr levels suggest that histone pr plays a role in metabolic signaling and could be implicated in metabolic diseases, although the specific genomic changes warrant further investigation. 140 Researchers suggest a possible association of histone bu and pr with conditions such as systemic lupus erythematosus, alcohol dependency, virus-induced cancer development and aberrant gene expression in oncological diseases. 141
Researchers have identified specific form of histone bu termed lysine isobutyrylation. Notably, this isobutyrylation stems from the catabolism of valine and the oxidation of branched-chain fatty acids, pointing to its extensive regulatory roles in epigenetics and cellular physiology. 142 Currently, the link between histone isobutyrylation and tumors is still unclear, with future research expected to explore this further.
Histone 2-hydroxyisobutyrylation
The Dai team identified a novel HPTMs, hib, through MS and then verified it through chemical and biochemical methods. The modification in question has been validated at 63 lysine hib sites within both human and murine histones. Investigations have determined that the histone hib mark is highly preserved across species and extensively dispersed, possesses elevated stoichiometry, and prompts notable alterations in structural configuration. These discoveries underscore the fundamental role of the histone hib mark in the governance of chromatin functionality. 8
Lysine hib, ac, and cr are positioned on separate residues within histones, each exhibiting distinct functional attributes divergent from those of lysine ac. 7 Research has pinpointed 63 instances of histone hib marks, a number that eclipses the identified histone lysine ac sites. Unlike the predominantly N-terminal tail localization of known cr and ac modifications, hib marks are discernible within both the N-terminal regions and the core globular domains of histones. Structurally, hib modifications diverge notably from lysine modifications such as ma, ac, or bu. Hib modifications do more than merely neutralize the positive charge of lysine; they also significantly expand its radius, indicating profound structural and functional implications. 8 Essentially, the hib modification is characterized by the addition of a hydroxyl group to the lysine residue. This confers upon the modified lysine the capacity to engage in hydrogen bonding with other molecular entities. Such a characteristic is of noteworthy importance, as it can substantially influence the modulation of protein functionality. 143 The genesis of hib is postulated to occur through an enzymatic process that utilizes a high-energy donor molecule, possibly sourced from 2-hydroxyisobutyrate, such as HibCoA, as a cofactor. This suggests a profound interconnection between cellular metabolism and epigenetic frameworks. Consequently, the hib pathway might provide a means for cells to adapt their epigenetic landscapes in accordance to fluctuations in the cellular concentration of HibCoA. The extent to which enzymes known to regulate lysine ac are involved in modulating hib remains an area of uncertainty. Initial in vitro assessments indicate that HDACs 1-3 have the potential to detach the 2-hydroxyisobutyryl moiety from its lysine counterpart under controlled experimental conditions; however, the validity of this activity within a cellular context awaits further verification. As a novel regulatory factor in genetic regulation, hib is closely associated with the metabolic state of the cell and plays a role in epigenetic regulation. 138 , 144 This paves the way for future research into the biological functions and regulatory mechanisms of hib.
Histone 2-hydroxybutyrylation
Initially delineated by Xie and colleagues in the year 2016, the histone modification termed bhb is dynamically modulated by the intracellular concentration of β-hydroxybutyrate. Despite this established correlation, the exact biochemical pathway through which β-hydroxybutyrate donates β-hydroxybutyryl groups to histones has yet to be fully expounded. 23 , 145 Investigations employing RNA sequencing techniques in conjunction with analyses utilizing the KEGG have demonstrated a robust correlation between the bhb of H3K9 and the upregulation of gene expression. When the concentration of β-hydroxybutyrate is elevated, bhb is highly expressed on histones. As an energy source during fasting states for the heart and brain, and in ketogenic diets, β-hydroxybutyrate plays a critical role. β-hydroxybutyryl-CoA, acting as a specific cofactor, catalyzes the formation of bhb in a concentration-dependent manner. 69 bhb is predominantly enriched at the promoters of active genes and is associated with genes that are upregulated in metabolic pathways responsive to starvation. 23 β-Hydroxybutyrate, a naturally occurring ketone body, is pivotal in the initiation and advancement of neurological disorders, underscoring its significant impact on neurobiological health. Through comprehensive research, scientists have discovered that the biological process of bhb can provide neuroprotection, effectively mitigating the toxic damage faced by neurons. Furthermore, this process also helps in preventing the degenerative changes in dopaminergic neurons among patients with AD and PD. 146 , 147 And p53, recognized as a pivotal tumor suppressor, undergoes modification through bhb at lysines 120, 319 and 370. Under conditions of starvation, researchers noted an elevation in β-hydroxybutyrate serum concentrations in mice, accompanied by a rise in p53 bhb. This process hampers the ac of p53, leading to the cessation of cellular proliferation and a reduction in programmed cell death. 148 This suggests to us the role of bhb in ketone metabolism and tumor management (Fig. 2 ) ( Table 1 ).

HPTMs and metabolism correlation diagram. Lysine acylation is a complex process interconnected with major metabolic pathways. Glucose, fatty acids, and amino acids serve as primary metabolic resources, producing a wealth of intermediate products within cells, such as lactate, succinyl-CoA, acetyl-CoA, and β-hydroxybutyrate. These intermediates supply acyl groups essential for the covalent modification of proteins. Notably, metabolites like cr, bu, and pr, as well as hib, predominantly arise from fatty acid oxidation and amino acid metabolism. Conversely, metabolites such as la, bhb, and succ primarily originate from glucose metabohighlighting a diverse metabolic sourcing for lysine acylation
Biological functions Of Hptms
Hptms and genome function, hptms in transcription.
During transcription, HPTMs of histones play a crucial role. These modifications often occur at specific genomic locations, notably enriching at genes, where their presence is associated with transcriptional activity, either positive or negative. 149 HPTMs is frequently linked to transcriptional engagement, being enriched at active promoters, enhancers, and other regions of chromatin that are readily accessible. Furthermore, it has been demonstrated that HPTMs directly augments the rate of transcription within in vitro settings. 150 , 151 Additionally, histone lysine residues are subject to modifications by various long-chain acyl groups. However, these types of modifications are generally observed with much lower prevalence compared to ac. 152 A quintessential example is histone crotonylation, 7 which was initially identified as a positive regulator of transcription. 153 It is of particular interest to note that cr, while typically implicated in gene activation, has also been associated with the repression of gene expression within yeast organisms. 154 Early studies indicated that the global levels of HPTMs are not related to transcriptional activity. 155 , 156 The consequence of HPTMs is highly contingent upon the specific site of occurrence and it is not likely to exert a direct influence on nucleosome architecture. 149 Although numerous HPTMs have been identified in association with transcription, direct evidence supporting their causal role in transcriptional regulation remains elusive. 157 Herein, we contemplate the role of specific HPTMs in the regulation of transcription, with particular emphasis on the position of the modified amino acids within the histones.
An array of specific modifications occurs at the histone tails, such as the H3K4ma3, which is concentrated near TSSs and aids in the recruitment of transcriptional machinery. This includes the transcription initiation factor TFIID subunit 4, which facilitates the expression of particular genes and is associated with the maintenance of transcriptional activity in a quiescent state in mammals. 158 , 159 Moreover, experiments in fruit flies and African clawed frogs have demonstrated that H3K4ma1 is essential for the memory of an active transcriptional state. 160 , 161 , 162 Particularly in the context of novel HPTMs, researchers have employed cell-free assays to demonstrate that histone la, akin to acetylation, can directly stimulate gene transcription. Experiments using l-lactyl-coenzyme A instead of acetyl-coenzyme A demonstrated p53-dependent, p300-mediated la of H3 and H4 and the corresponding transcriptional effects. The direct mediation of transcription by histone la was confirmed using recombinant chromatin with lysine-to-arginine mutated core histones. Histone la is not essential for the induction or repression of pro-inflammatory genes but is employed to initiate the expression of homeostatic genes traditionally associated with M2-like macrophages. The aerobic glycolytic switch that occurs during M1 polarization triggers a “lactate timer” that induces M2-like characteristics at a later stage through epigenetic mechanisms, potentially aiding in repairing collateral damage suffered by the host during infection. 21 Furthermore, the distribution of the epigenetic mark H3K9bhb on histone H3 correlates with alterations in gene expression, capable of reshaping the chromatin landscape and transcriptional responses in brain cells. Histone cr plays a dual regulatory role in gene expression, acting as both a transcriptional activator and repressor, depending on its location and associated genes. Sirt3 was found to reduce the expression levels of Ptk2, Tshz3, and Wapal and decrease the enrichment of histone cr at the promoters of these target genes, indicating that histone cr may serve as a positive regulatory factor for the expression of these genes. 71 For instance, one study discovered that H3K9cr peaks at pro-growth genes led to gene repression, suggesting that H3K9cr is associated with the transcriptional suppression of pro-growth genes. 154
In addition to the modifications occurring on histone tails, modifications to core histones exert significant influence on gene expression. Lateral Surface HPTMs can directly affect the binding affinity between histones and DNA, as well as the rate at which DNA unwinds and rewinds. 55 , 56 Modifications on the lateral chains of histones can modulate the accessibility of DNA within nucleosomes and promote nucleosome mobility. This particular modification accelerates the rate of local DNA unwinding and induces spontaneous local conformational changes, phenomena colloquially termed as “DNA breathing.” Consequently, this fosters an enhanced affinity for transcription factor binding in vitro experiments. 163 , 164 HPTMs around the symmetry axis decrease the overall affinity of DNA for the histone octamer, thereby reducing nucleosome stability. 57 , 58 , 124 , 165 These core modifications exert their effects through reader or effector proteins.
The direct impact of multiple histone tail HPTMs on nucleosome stability and chromatin architecture is limited, and they typically exert biological effects through the recruitment of binding proteins, or effectors. Conversely, HPTMs that are located within the core domain of the histone octamer are inclined to exert a more direct impact on nucleosome structure and functionality. These intrinsic alterations have the potential to impinge upon chromatin-dependent processes, and this can occur independently of the presence of specific reader proteins. These insights offer a significant perspective on the dynamic modulation of chromatin structure and its impact on the regulation of gene expression, revealing a complex and nuanced interplay between histone modifications and transcriptional regulation. Through the study of these mechanisms, we can gain a more profound understanding of the expression and regulation of information within the cell, as well as unveil potential new strategies for the treatment of diseases, particularly cancer and genetic disorders.
HPTMs in recombination
In eukaryotic organisms, meiotic recombination and V(D)J recombination are two critical DNA recombination processes that play a central role in genetic diversity and the development of the immune system. Both processes involve specific HPTMs.
Meiotic recombination frequently takes place at genomic locales known as hotspots, which are characterized by an abundance of open chromatin mark. These epigenetic adornments are intricately arranged by PRDM9—a zinc finger DNA-binding protein with testis-specific expression. PRDM9, in synergy with the lymphoid-specific helicase HELLS, constitutes a vanguard complex. This coalition functions to render the chromatin more accessible, thereby enabling the facilitation of meiotic recombination. 166 Studies have revealed that H4K8la is closely associated with recombination hotspots, which involve mechanisms that process DSBs, such as SPO11, DMC1, RAD51, and RPA2. Moreover, H4K8la has also been detected at meiosis-specific cohesion sites (marked by RAD21L and REC8) flanking the recombination hotspots. 167 While PRDM9 plays a significant role in delineating the landscape for meiotic recombination, its presence is not indispensable for the recombination process to occur, as evidenced in the context of rats. 168 In the majority of vertebrates lacking PRDM9, recombination activity shifts toward other open chromatin structures, such as the promoters of active genes. From a topological perspective, the loop anchors of topologically associating domains (TADs) are enriched with H3K4ma3 and contain multiple PRDM9 binding sites. These features may explain why loop anchors can serve as hotspots for meiotic recombination. 169 , 170
The process of V(D)J recombination relies on the functionality of proteins produced by the Recombination Activating Genes, which assemble into a complex consisting of RAG1 and RAG2 subunits. This complex is characterized by its recombinase activity and its ability to bind to the highly conserved recombination signal sequences that border the V, D, and J gene segments. These genomic regions associated with V(D)J recombination are marked by active histone modifications. The interaction of the plant homeodomain within RAG2 with H3K4ma3 triggers a conformational alteration in RAG1. This modification is pivotal in enhancing the catalytic efficiency necessary for recombination. 171
Overall, H3K4ma3 plays a pivotal role in both types of recombination. In meiotic recombination, it interacts with PRDM9, influencing chromatin accessibility and the selection of recombination sites; in V(D)J recombination, HPTMs is directly involved in the activation and functional deployment of the RAG complex, illustrating its guiding role in immune diversity. These findings highlight the dual function of HPTMs in DNA recombination: modulating transcription and directly impacting the recombination mechanism.
HPTMs in DNA repair
HPTMs are critically involved in the cellular mechanisms addressing DNA damage response and repair, particularly when confronting double-strand breaks (DSBs), which represent the most severe form of DNA damage. Genomic integrity is continually challenged by DNA damage, which is a hallmark of cancer. 172 The propagation of γH2AX coincides with the boundaries of TADs and may be facilitated by the process of loop extrusion mediated by cohesin, aiding in the spread of γH2AX from the site of the double-strand break. 173 , 174 γH2AX provides a platform for the recruitment of DNA damage signaling factors, which initiate ubiquitination of H1 and H2A histones mediated by the ubiquitin ligases RNF8 and RNF168, triggering downstream repair processes. 174 , 175 The functional engagement of these signaling entities is pivotal in dictating the subsequent choice of DNA repair pathways. Double-strand breaks are predominantly mended through two principal mechanisms: Homologous Recombination (HR) and Non-Homologous End Joining. 176 HPTMs modulate the balance between the two repair pathways by influencing the binding and activity of 53BP1 and BRCA1. The affinity of different reader domains for cell cycle-regulated and DNA damage-dependent HPTMs collectively and distinctively determines the choice of double-strand break repair pathway. There, they act to inhibit transcription and promote the recruitment of DNA repair factors, thereby facilitating the repair process. 177 , 178 Furthermore, research indicates that H3K9 la is significantly enriched in the LUC7L2 promoter, activating LUC7L2 transcription to enhance its expression. LUC7L2 mediates the retention of intron 7 in MLH1, thereby reducing MLH1 expression and inhibiting mismatch repair, ultimately leading to TMZ resistance in glioblastoma multiforme (0GBM). 179 The orchestration of DNA repair and transcriptional processes is further regulated by the intricate interplay of HPTMs. These HPTMs reveal how chromatin states influence a cell’s capacity to respond to and efficiently repair DNA damage by affecting chromatin structure and function during the DNA damage response and repair processes. Histone modifications play a crucial role in maintaining genomic integrity and preventing the development of cancer.
HPTMs in replication
HPTMs play a pivotal role in regulating DNA replication. The modifications, particularly ac and ma on specific histone lysine residues, are essential for the setup and activation of replication origins and influence the overall chromatin structure. These alterations facilitate the assembly of necessary replication complexes, impacting the initiation and efficiency of DNA replication. Additionally, HPTMs are involved in defining the replication timing across the genome, which is crucial for maintaining genome stability and organization. The dynamic relationship between DNA replication and HPTMs suggests that changes in replication timing can alter histone modifications, thereby affecting genomic architecture and compartmentalization within the nucleus. This highlights the significance of a regulated replication process in preserving the integrity and functionality of the genome 180 , 181 , 182 , 183 , 184 (Fig. 3 ).

T he crucial role of HPTMs in genome function. This figure illustrates the crucial roles of HPTMs in regulating key genomic functions, such as transcription, replication, DNA repair, and recombination
HPTMs and cancer metabolism
HPTMs are intimately connected to metabolism. Through metabolic pathways, glucose is broken down into pyruvate and lactate, both of which are associated with specific histone modifications. Lactate is directly linked to histone la, while pyruvate is further converted into acetyl coenzyme A, a substrate for various acylation modifications, including bu. Fatty acids undergo β-oxidation within mitochondria, leading to the production of intermediates like Ac-CoA, which are not only pivotal for energy generation but also fundamental to histone modification processes. For instance, Ac-CoA can directly contribute to histone acetylation. Succinyl coenzyme A and hydroxybutyryl coenzyme A are two other acyl-CoA molecules, which can result in histone succ, bhb, and hib, respectively. The oxidation of long-chain fatty acids also produces various long-chain acyl-CoAs, such as propionyl coenzyme A, which is associated with histone pr. Moreover, amino acids like lysine and tryptophan can be metabolized into their corresponding acyl-CoA derivatives. These acyl-CoA molecules subsequently react with histone lysine residues, leading to modifications such as cr. These processes fundamentally represent the interplay between cellular metabolism and epigenetic modifications, reflecting how cells adjust gene expression and protein function in response to varying metabolic states.
The connection between epigenetics and metabolism is bidirectional, encompassing both research into how HPTMs control the expression of metabolic genes, and investigations into how metabolic pathways influence these newly discovered HPTMs. This bidirectional interaction reveals the complex interplay between epigenetics and metabolic processes, enhancing our understanding of the regulatory mechanisms within organisms. Recent studies reveal that histone modifications, including la, cr, and succ, are intricately linked to cancer metabolism. For instance, histone la bridges cellular metabolism and gene expression, potentially revealing its disease mechanisms. A recent study conducted an analysis of differentially lactylated proteins across various groups, revealing their involvement in a wide range of biological functions. These functions include amino acid and lipoprotein metabolism, as well as the synthesis of ribosomal proteins. This analysis confirmed the close association between histone la and HCC. Furthermore, the investigation confirmed the levels of la on two proteins associated with tumors, namely USP14 and ABCF1, establishing a solid foundation for further exploration of their roles in HCC pathogenesis. 185 Additionally, the roles of histone cit in NET formation and SUMOylation in cell regulation provide promising directions for cancer therapy development. Furthermore, emerging modifications such as bu and bhb underscore the significance of metabolic pathways in cancer progression, offering new therapeutic possibilities (Fig. 4 ).

Mechanisms linking novel HPTMs to cancer development through metabolic pathways. The p300 protein possesses key lysine 2-hydroxyisobutyryltransferase activity, playing a crucial role in regulating glycolysis. Aspirin, by specifically hydroxyisobutyrylating at certain sites, inhibits the 2-hydroxyisobutyrylation of the key glycolytic enzyme ENO1, thereby reducing its activity and consequently inhibiting the growth of tumor cells. Additionally, the rate of aerobic glycolysis in tumor cells is increased, accompanied by an increase in histone lactylation, which transcriptionally supports the expression of c-Myc. As a critical transcription factor, c-Myc further upregulates the expression of SRSF10, promoting the selective splicing of MDM4 and Bcl-x in breast cancer cells, thus affecting the growth and survival of cancer cells. On the other hand, aspirin significantly reduces the succ levels of PGAM1 in liver cancer cells, thereby inhibiting glycolysis. Concurrently, 2-hydroxybutyrate induces 2-hydroxyisobutyrylation modifications of p53 at lysine residues 120, 319, and 370. These modifications lead to reduced acetylation levels of p53, subsequently downregulating the expression of downstream genes p21 and PUMA, ultimately resulting in reduced growth and apoptosis of cancer cells
Glucose metabolism
Cancer cells primarily rely on glycolysis for energy production. HPTMs, particularly histone la and hib, have the potential to impact cancer therapy by influencing this glycolytic pathway. Some investigations have revealed that p300 possesses 2-hydroxymethylisobutyryltransferase activity, and it modulates cellular glycolysis by amplifying the levels of hib at specific sites in P1. Empirical data indicate that a deficiency in p300 diminishes the activity of enzymes involved in glycolysis, underscoring the potential of modulating hib levels by inhibiting p300 to constrain tumor growth. 186 The study suggests that in cells affected by neuroendocrine, prostate, or lung cancer, mitochondria are often more fragmented and show a reduced membrane potential, primarily depending on glycolysis for their energy production. Additionally, the interaction between Numb and Parkin promotes mitophagy, crucial for maintaining mitochondrial quality. The Numb/Parkin pathway acts as a critical metabolic regulator and emerges as a potential therapeutic target in oncology. 187 Aspirin notably decreases overall succ levels in liver cancer cells, including the succ of phosphoglycerate mutase 1 (PGAM1), thereby curtailing the glycolytic process. 188 For instance, research conducted by Pandkar and their team indicated that decreasing glycolytic activity, especially by inhibiting histone la, significantly reduces the expression of the c-Myc gene, which in turn hinders the advancement of breast cancer. 189
Lipid metabolism
Researchers have found that HPTMs, specifically bhb and bu, are closely associated with ketone body metabolism. In dietary screening studies conducted on spontaneous animal models of CRC, researchers discovered that ketogenic diets possess significant tumor-suppressing effects. This anti-tumor efficacy is replicated through the ketone body β-hydroxybutyrate. β-hydroxybutyrate acts by interacting with the surface receptor Hcar2, subsequently inducing the activation of the transcriptional regulator Hopx. This activation leads to alterations in gene expression patterns, effectively inhibiting the proliferation of colonic crypt cells and significantly suppressing intestinal tumor growth. 190 Another research indicated that inhibiting the initiation of ketogenesis within the tumor microenvironment is a critical factor in the progression of colorectal cancer. Ketogenic diet has been identified as a key modulator of the tumor microenvironment, capable of diminishing the accumulation of immunosuppressive cells within tumors, enhancing the infiltration of natural killer cells and cytotoxic T cells, and amplifying the anticancer efficacy of PD-1. 191 Additionally, another study highlighted the positive impact of a ketogenic diet on cancer treatment by restricting glucose and upregulating histone bu, particularly in relation to breast cancer. 192 One research indicates that β-hydroxybutyrate triggers hib on p53 protein at lysine residues 120, 319, and 370. This cascade of precise molecular regulatory actions leads to a reduction in the ac levels of p53, which in turn permeates through the downstream gene network, significantly suppressing the expression of the genes p21 and PUMA. Ultimately, this series of intracellular signaling events acts in concert to effectively inhibit the proliferation of cancer cells and induces them towards the path of apoptosis, thereby playing a pivotal role in anti-cancer strategies. 148 However, the possible benefits of a ketogenic diet in cancer treatment, but its definitive effectiveness is still uncertain. 193 Studies have shown that after long-term treatment, the accumulation of β-hydroxybutyrate and glucose restriction did not significantly affect the levels of bu or ac of histone H3 in cancer cells. Researchers believe that the metabolic plasticity of cancer cells, through limiting glucose and the enrichment of histone modifications, can mitigate or neutralize the effects of long-term metabolic reprogramming. This provides new insights into the controversial mechanism of action of ketogenic diets in clinical trials. 192
Glutamine metabolism
Cancer cells rely extensively on glutamine to fuel their growth, utilizing it in processes like lipid synthesis, the tricarboxylic acid cycle, and for generating amino acids and nucleotides. This ‘addiction’ to glutamine in cancer cells underlines its critical role in the metabolism of tumor cells. Glutamine, absorbed from plasma through different amino acid transporters and transformed into glutamate by mitochondrial glutaminase, plays a pivotal role in glutaminolysis. This process is considered a primary target in the development of cancer therapeutics. 194 Existing research has outlined the importance of histone ac and glutamine metabolism in cancer, yet the impact of other HPTMs in this context is still not fully understood. Future studies aim to explore their interaction in cancer treatment, potentially identifying new therapy targets. Furthermore, some studies have pinpointed that HPTMs, particularly hib, have a pronounced association with carbohydrate metabolism. 195
HPTMs and genome topology
Within the genomes of animals and plants, the chromatin structure is organized into two distinct compartments: A (euchromatin) and B (heterochromatin), each characterized by structural differences. 196 These compartments are further delineated into topologically associating domains, or TADs. Within the elegant model organism C. elegans, the demethylase of H4K20ma2 stands as one of the key complexes that catalyze the emergence of these TADs. 197 H3K9 ma plays a crucial role in the compartmentalization of the 3D genome. The deliberate localization of the enzyme that catalyzes the formation of H3K9ma3 to designated sites within human cells, achieved through dCas9-mediated guidance, fosters the tethering of chromatin to HP1α condensates. This strategic recruitment induces a substantial reconfiguration of the chromatin compartments, exemplifying the capacity of targeted histone modifications to alter nuclear architecture. 198 , 199 , 200 Moreover, H3K27ma3 is equally pivotal in the spatial organization of the genome. It is essential for the formation of chromatin regions known as Polycomb-associated domains, or PADs, through the recruitment of PRC1, yet it is not requisite for their maintenance. 201 These findings indicate that the genesis of TADs may be delicately orchestrated through the synergistic interactions between DNA and histone modifications. Collectively, HPTMs are deeply entwined with the governance of DNA replication, repair, transcription, and the three-dimensional organization of the genome, serving as pivotal elements in ensuring genomic stability and functionality.
The role of novel Hptms in diseases
Histone modifications regulate a myriad of physiological mechanisms. Thus, it is unsurprising that their dysregulation is implicated in various complications and diseases. Yet, among the array of conditions bearing the hallmark of epigenetic aberrations, cancer stands as the most thoroughly investigated and distinctly characterized ‘epigenetic disease.’ Feinberg, Ohlsson, and Henikoff have posited that the trajectory of tumorigenesis advances through a tripartite progression: (a) the initial epigenetic disarray within stem/progenitor cells, orchestrated by the aberrant regulation of tumor progenitor genes; (b) the subsequent genetic perturbations affecting tumor suppressor genes and oncogenes; and (c) a phase of compounded genetic and epigenetic volatility, culminating in an accelerated pace of tumor evolution. 202 DNA ma and various histone modifications play a role in this process, potentially silencing tumor suppressor genes or compromising genomic stability, ultimately resulting in cancer 203 , 204 (Fig. 5 , 6 ).

Schematic of HPTMs and the association with cancers. a The nine types of histone modifications was added by ‘writers’ and removed by ‘erasers’. These modifications are crucial for the regulation of gene expression. b The connections between different modification sites and various types of cancer

The role of novel HPTMs and related molecular markers in progression and prognosis of various cancer types in the human body. In the context of MI, monocytes first undergo metabolic reprogramming toward glycolysis, becoming dysregulated and committing to increased lactate production. The lactate is then taken up by MCT1 into monocytes, where it accumulates within the cell as a substrate for histone la, leading to histone la-mediated activation and expression of reparative genes including Lrg1, Vegf-a, and IL-10. These effects of monocyte gene induction enable monocytes to exert dual anti-inflammatory and proangiogenic activities, enhancing cardiac repair. Moreover, an increase in calcium ion concentration is the stimulus for PAD4 activation, which brings citrullinated histones, forming NETs. NETs are composed of DNA, histones, and antimicrobial proteins and are released to the extracellular space. These NETs elicit proinflammatory responses, myocardium- and endothelium-damaging effects, and interaction with platelets to elicit TGFβ release that may propagate a myofibroblast-driven fibrotic response. Indeed, NET formation and fibrosis are enhanced in the context of MI, and such responses are absent in mice deficient in PAD4, which have less fibrosis and a favorable outcome in terms of cardiac function
Ocular melanoma
Yu et al. discovered that histone 1a can activate the m6A reader protein YTHDF2, which is capable of recognizing m6A modifications on PER1 and TP53 mRNA, thereby promoting their degradation and accelerating the progression of ocular melanoma. 205 , 206 Utilizing western blot analysis, researchers uncovered a correlation between increased histone la and adverse outcomes in ocular melanoma patients. A notable feature of many ocular melanomas is the elevated histone la, which might contribute to the tumorigenesis of ocular melanoma. 205
Bladder cancer
a study suggests that the circXRN2-Hippo signaling pathway plays a role in controlling tumor advancement by suppressing H3K18la and regulating the expression of LCN2 in cases of human bladder cancer. 207
Acute myeloid leukemia (AML)
Clinically, researchers have found a positive correlation between the accumulation of lactate in the bone marrow of AML patients and the expression of STAT5 and PD-L1. This correlation is attributed to the overexpression of STAT5 promoting the nuclear translocation of E3BP, thereby activating the promoters of glycolytic genes, which in turn stimulates histone la and induces the transcription of PD-L1. This suggests that patients exhibiting high expression and subsequent production of lactate may benefit more from immunotherapies targeting the PD-1/PD-L1 axis. 208
Liu and colleagues discovered pr of histone H3 in mammalian cells. Using specific antibodies of pr, they further detected pr in the leukemia cell line U937. The team observed a significant reduction in H3K23 pr within these leukemia cells. Based on these findings, it is conjectured that histone pr might have implications in the pathogenesis of leukemia and potentially serve as a diagnostic marker for therapeutic interventions in leukemia. 65
Clear cell renal cell carcinoma (ccRCC)
Studies have indicated that the expression of histone la is closely associated with cancer prognosis. Particularly in ccRCC, the inactivation of the VHL gene directly increases the expression of histone la, which correlates with poor patient outcomes. Furthermore, the expression of histone la, induced by the inactive state of VHL, further accelerates the development of ccRCC by promoting the transcription of PDGFRβ. In turn, the activation of PDGFRβ also stimulates the transcription of histone la, creating a positive feedback loop that hastens tumor progression. These findings offer significant insights into the molecular mechanisms of ccRCC and may inform the development of therapeutic strategies. 209
Colorectal cancer(CRC)
At the same time, research indicates that the activity of histone la is associated with tumor metastasis and aggressiveness. Wang and colleagues found that lipopolysaccharides from intestinal bacteria can upregulate the expression of LINC00152 in colorectal and breast cancers, 210 affecting the tumor microenvironment. This upregulation is achieved through an increase in histone la activity induced by lipopolysaccharides and a reduction in the binding affinity of the repressive factor YY1, subsequently promoting LINC00152 expression. Further studies have revealed that overexpression of LINC00152 enhances the migratory and invasive capabilities of cancer cells, highlighting its pivotal role in cancer progression. 211
The research conducted by Qu and colleagues has revealed that under hypoxic conditions, HIF-300α promotes the development of cancer cells by recruiting p300/CBP to increase Autotaxin(ATX) expression, particularly through the crotonylation of H3 in colon cancer cells, thereby activating ATX. Moreover, the increase of histone crotonylation under normoxic conditions can also initiate the expression of ATX. 212 In parallel, Liao’s team has established a link between histone crotonylation and DNA damage activity in CRC patients through bioinformatics and Western blot techniques. These findings further highlight the significant roles of histone crotonylation and the cancer therapeutic target ATX in cancer progression. 213
Hou and his team evaluated the association between histone cr and tumor staging as well as diagnostic outcomes. Their investigation showed a notable increase in H2BK12 cr within the peripheral blood mononuclear cells from patients with CRC. Through ROC curve analysis, it was discerned that utilizing H2BK12cr levels as a diagnostic criterion significantly surpassed conventional carcinoembryonic antigen tests in terms of simplicity and efficacy, thereby highlighting its considerable potential as a tumor biomarker. 214 Challenging previous views, Liu et al.‘s recent research reveals a new role for histone cr beyond gene activation. Their study suggests that cr at histone H3 lysine 27 (H3K27cr) primarily acts as a suppressor rather than an enhancer of gene transcription. They found that the YEATS domain within the GAS41-SIN3A-HDAC1 complex uniquely recognizes H3K27cr in chromatin. Remarkably, the transcription factor associated with proto-oncogenes, MYC, recruits this complex to repress gene expression within chromatin subsequently. Furthermore, their experiments on mice revealed that either the knockdown of GAS41 or the deletion of H3K27cr binding can contribute to the inhibition of tumor growth. This sheds light on the novel role that histone cr plays in tumorigenesis. 215
Hepatitis B virus-related hepatocellular carcinoma(HCC)
A study involving patients with hepatitis B virus-related HCC revealed an increase in histone H3 cit levels, which is closely associated with Beclin1 mRNA expression, vascular invasion, and serum AFP levels, reflecting a critical aspect of liver cancer progression. 216
Significantly, HAT1, conventionally acknowledged as a HAT, has been additionally recognized for its succinyltransferase activity. In their quantitative proteomic analysis of HepG2 cancer cells, Yang and colleagues discovered that HAT1 coordinates the succinylation of a wide array of proteins, including histones and non-histone proteins. Furthermore, their investigations revealed that HAT1 is capable of H3K122succ, subsequently promoting favorable gene expression patterns within cancer cells. Clinically, elevated HAT1 levels have been observed in various cancerous tissues, such as HCC, pancreatic carcinoma, and cholangiocarcinoma. Therefore, it can be inferred that the succinyltransferase function of HAT1, coupled with the succ of PGAM1 mediated by HAT1, stands as a critical mechanism driving tumor progression. 217
In 2016, Zhao et al. revealed that 2-hydroxybutyric acid can act as a foundation for the bhb of histones, activating gene expression. Intrinsically, β-hydroxybutyrate is a primary component of ketone bodies. This significant elevation in bhb levels effectively links gene expression to ketone body metabolism. 218 Tumor cells demand substantial energy for growth, and in contrast to normal cells, this energy predominantly comes from glycolysis. 219 Existing studies indicate that β-hydroxybutyrate or β-hydroxybutyrate-induced HPTMs may have pivotal roles in the onset and treatment of tumors. Scientists have observed that the build-up of β-hydroxybutyrate followed by the elevated presence of H3K9bhb in living organisms, mediated by MTA2, can initiate a cascade response fostering the progression of HCC. Additionally, it has been noted that the upregulation of the genes JMJD6, GREB3, GTPBP4, NPM1 and TIMM23 can affect the prognosis of HCC patients. 220
Gastric cancer
In a distinct study, Zheng and colleagues provided persuasive findings showing that PADI2 plays a crucial role in promoting angiogenesis, cellular growth, movement, and influencing the tumor immune environment. This is achieved by amplifying the expression of CXCR2, KRT14 and TNF-α, thereby facilitating the onset of gastric cancer. 221 Additionally, another study vividly showcased the close association between group mono-leuco-citrullination and IPO-38, emphasizing the potential of the latter as a biomarker for early gastric cancer detection. 222
Prostate cancer(PCa)
In PCa research, Wang and colleagues discovered the crucial function of PADI2 in cell viability and cell cycle advancement. Their findings indicate that PADI2 facilitates the growth of prostate cancer cells. 223 In a separate investigation, it was determined that PAD2 influences breast cancer by modulating histone cit. 224
The research led by Xu and his team has accentuated the role of histone cr in PCa, uncovering that levels of histone cr are significantly elevated in PCa tissues compared to adjacent normal tissues and are closely associated with the severity of the disease. Through immunohistochemical analysis of 72 PCa patient samples and laboratory assays on three human PCa cell lines—including quantification by Western blot, as well as assessments of cell proliferation, migration, and invasion—the study unveiled potential mechanisms of histone cr in cancer progression. These findings suggest a significant correlation between the expression of histone cr and the clinical staging and grading of PCa, indicating its potential as both a prognostic marker and a therapeutic target. 225
Pancreatic ductal adenocarcinoma
Histone succ is modulated by KAT2A, an enzyme exhibiting both lysine acetyltransferase and succinyltransferase functionalities. Investigations have revealed that KAT2A is not only abundantly manifested in human pancreatic ductal adenocarcinoma but also displays a positive correlation with the progressed stages of pancreatic ductal adenocarcinoma and a diminished patient survival rate. It is demonstrated that KAT2A augments the migration and invasiveness of pancreatic ductal adenocarcinoma cells through the regulation of 14-3-3ζ and β-catenin expression. 129 The nuclear-localized α-ketoglutarate dehydrogenase complex in human cells has been identified to associate with KAT2A at the gene’s promoter region. KAT2A has the capability to execute succ on lysine 79 of histone H3. Acting as a succinyltransferase, KAT2A induces succ of histone H79 at lysine 3, predominantly occurring in proximity to the gene’s transcriptional initiation site. Experimental findings suggest that by obstructing the nuclear entry of the α-ketoglutarate dehydrogenase complex or suppressing the expression of KAT2A (Tyr645Ala), one can diminish gene expression, which consequently reduces the proliferation of tumor cells and overall tumor expansion. 126 Concurrently, studies have shown that genomic instability is a hallmark of cancer. 226 This association between histone succ and DNA damage has been underscored by several studies. 227 To sum up, research has demonstrated that histone succ, regulated by KAT2A or HAT1, is pivotal in governing gene expression. This regulation significantly contributes to the growth, advancement, and metastasis of cancer cells.
Lu and his team, utilizing liquid chromatography and LC-MS/MS techniques, have for the first time identified histone hib sites in patients with pancreatic cancer. They discovered that histone modifications involving hib affect key metabolic pathways such as glycolysis, gluconeogenesis, and the TCA cycle, highlighting the significance of hib in the metabolism of pancreatic cancer. Moreover, the inhibition of Tip60 significantly suppresses the growth, migration, and invasion of pancreatic cancer, which correlates with the downregulation of hib. These results suggest that histone hib modifications are closely linked to the progression of pancreatic cancer. 228 Additionally, predictive analyses using IPA software have revealed the role of actin cytoskeleton regulatory pathways in the development of oral squamous cell carcinoma, pointing to the regulation of actin assembly and stability as potential key mechanisms in cancer progression. 229
Non-small cell lung cancer(NSCLC)
Furthermore, Tanikawa and his team observed in NSCLC that the levels of histone H4R3 cit are inversely proportional to p53 expression and tumor size, and that the p53 pathway, mediated by PADI4, significantly affects tumorigenesis. 230
In essence, the citrullination-induced NET seems to ominously forecast a grim prognosis in the realm of cancer. An extensive analysis with long-term patient follow-up in cases of cancer-related VTE revealed a significant correlation: the cit status of histone H3 was strongly associated with VTE. This discovery strongly underscores the vital importance of NETs in the progression of VTE within oncogenic settings. 231 Similarly, another investigative endeavor revealed an intriguing observation: elevated plasma concentrations of the NET biomarker, citrullinated histone H3 (H3cit), were concomitant with the onset of VTE in individuals diagnosed with pancreatic and lung cancers. 232 In preliminary studies, researchers have observed a marked elevation in the levels of citrullinated H3cit in the plasma of patients with advanced cancer, and this elevation is tightly associated with the patients’ prognosis. The significant increase in H3cit levels nearly doubled the short-term mortality risk for patients, strongly highlighting its immense potential as a prognostic biomarker for cancer. 233
Prolactinomas
For instance, DeVore and colleagues discovered that in prolactinomas, the activity of PAD2 and PAD4 and the levels of histone cit are elevated, promoting tumor growth. This process facilitates tumor development and proliferation through the downregulation of microRNAs, such as let-7c-2 and 29c. 234
In another investigation, core histone lysine bu sites, such as H3K18, H3K23, H3K79 and H4K77, were identified in esophageal squamous cell carcinoma cell lines. 12
It has been shown that histone SUMOylation can function as a transcriptional repressor. This repression is primarily achieved either by disrupting histone ac or by interfering with the ubiquitination of H2BK123 through Rad6 and Bre1. 131 The SUMO system comprises the activation enzyme E1, the conjugation enzyme E2 and the ligating enzyme E3. The E1 and E2 enzymes are limited in variety—E1 encompasses SAE1 and SAE2, while UBC9 is the sole E2 enzyme. In contrast, E3 enzymes possess a more extensive range owing to their diverse specificities, which facilitate binding to various substrates. 235 Existing scholarly works indicate that Ubc9 holds a key role in regulating processes such as the cell cycle, cell growth, mitotic division, programmed cell death and DNA restoration. The direct correlation of Ubc9’s role in tumors histone SUMOylation remains somewhat nebulous. Nevertheless, the SUMOylation of multiple proteins is ascertained to significantly influence tumorigenesis. Significantly, increased levels of Ubc9 have been detected in a range of cancers such as lung, 236 prostate, 237 ovarian, 238 bladder cancer 239 and melanoma. 240 One research demonstrates that inhibiting UBC9 curtails tumor development in mouse xenograft models, while the depletion of Ubc9 fosters STAT4-mediated macrophage activation. This activation, coupled with enhanced macrophage-CD8 + T-cell communication, serves to thwart tumor progression. 237 The connection between SUMOylation and the onset of cancer is widely acknowledged, and continued investigation in this area is expected.
P53, recognized as a pivotal tumor suppressor, undergoes modification through bhb at lysines 120, 319 and 370. Under conditions of starvation, researchers noted an elevation in 2-hydroxybutyric acid serum concentrations in mice, accompanied by a rise in p53 bhb. This process hampers the ac of p53, leading to the cessation of cellular proliferation and a reduction in programmed cell death. 148 This suggests to us the role of bhb in ketone metabolism and tumor management.
The role of histone modifications in cancer is multifaceted: different types of cancer, and even individual cases within the same cancer type, may exhibit distinct patterns of histone modifications. This underscores the highly heterogeneous nature of cancer as a disease, necessitating further research to better harness the potential of histone modifications in treatment and prognostic applications.
Infectious diseases
Infectious diseases are complex biological processes closely associated with autoimmunity and inflammation. During the course of infection, microbes can induce epigenetic modifications, such as the suppression or promotion of expression of various inflammasomes. 241 , 242 Numerous reports have mentioned the pivotal role of HPTMs such as phosphorylation, acetylation, or methylation in the pathogenesis of viral infections—including Herpes Simplex Virus, Hepatitis B Virus, and Human Immunodeficiency Virus (HIV)—as well as in sepsis models. However, research on the mechanisms related to novel HPTMs in the context of infectious bacterial or viral diseases remains scant. 243 , 244 , 245 Research has found that all subjects, including healthy volunteers, express H3K18la; however, individuals with septic shock exhibit the highest levels. This suggests that H3K18la may reflect the severity of critical illness and the presence of infection. H3K18la could potentially regulate the anti-inflammatory function of macrophages in sepsis by promoting the expression of inflammatory cytokines and the overexpression of Arg1. 246
Studies have demonstrated that by augmenting the expression of ACSS2 (Acyl-CoA Synthetase Short-chain Family Member 2), histone cr can be induced, thereby reactivating latent HIV. Pharmacological inhibition or siRNA-mediated knockdown of ACSS2 can reduce HIV replication and activation. Moreover, when used in conjunction with protein kinase C agonists or histone deacetylase inhibitors, ACSS2 can efficiently reactivate latent HIV. In simian models of HIV, an increase in ACSS2 expression correlates with alterations in fatty acid metabolism. This research links ACSS2 with HIV latency and offers a potential new target for HIV eradication strategies. 116 The study by Jiang et al. suggests that histone cr is an epigenetic modification present on the long terminal repeats of HIV, which modulates the transcription and latency of the virus. Inhibition of cr can prevent the reactivation of latent HIV, indicating that the suppression of cr could contribute to the maintenance of HIV latency, offering a potential target for controlling and reducing the incidence of AIDS. 116
Cardiovascular diseases
Recent research findings indicate that histone la is critical for the preservation of cardiac sarcomere structure and function. This modification is specifically directed at the α-myosin heavy chain (α-MHC), which is a major contractile protein in the myocardium. La strengthens the interaction between α-MHC and titin 247 —a key sarcomeric protein that supports the elasticity and integrity of muscle tissue—thereby la contributes to maintaining myocardial contractile function. Scholars hypothesize that augmenting α-MHC la could emerge as an innovative therapeutic approach for combating heart failure. This hypothesis suggests that focusing on α-MHC la presents a potential new avenue for treatment strategies in heart failure management. Myocardial infarction triggers a complex inflammatory response, which is crucial for the control of acute damage and subsequent cardiac repair. 248 The initial cellular response encompasses a systemic emergency hematopoiesis reaction coupled with the swift mobilization of neutrophils and monocytes to the site of action. 249 , 250 A sustained excessive inflammatory response can exacerbate myocardial damage and cardiac dysfunction. The prompt initiation of reparative signaling within monocytes and macrophages is critical for the expeditious re-establishment of immune equilibrium and the commencement of the healing process post-myocardial infarction. 251 , 252 Research suggests that histone la can influence the anti-inflammatory and pro-angiogenic behavior of monocyte-macrophage cells, thereby promoting the transcription of genes associated with repair. This contributes to a healing milieu and enhances cardiac performance post-myocardial infarction. These insights uncover the pivotal function of IL-1β-induced recruitment of GCN5 (General Control Non-repressed 5) to histone H3K18la, shedding light on its possible role as a regulatory precursor for monocyte histone la and the subsequent expression of genes involved in repair following myocardial infarction. 253
Vascular diseases are associated with changes in PAD activity and citrullinated proteins. 254 , 255 , 256 , 257 , 258 , 259 , 260 , 261 In cardiomyocytes and fibroblasts, PAD2 and PAD4 have been identified as the predominant isoforms. 262 HPTMs of myofibrillar proteins in cardiomyocytes, such as cit, phosphorylation, oxidation, and ac, can lead to alterations in the structure and function of these proteins, resulting in diminished cardiac contractility. 262 In ischemic heart diseases, there is a marked increase in the cit of myosin heavy chains, and in patients with heart failure, there is an elevated cit of cardiac contractile proteins by PAD2. 262 PAD4 induces the formation of NETs, activates platelets, and promotes the secretion of transforming growth factor-beta (TGF-β), ultimately contributing to cardiac fibrosis. 263 , 264 Moreover, the absence of PAD4 has been shown to confer cardioprotection in mice. 263 Extracellular DNA or NETs adversely affect cardiac function following acute myocardial infarction injury. 265 Chromatin remodeling is crucial for controlling gene expression and cardiac growth in response to acute and chronic stimuli. 266 , 267 Moreover, in various models of deep vein thrombosis, it has been demonstrated that NETs can instigate coagulation. NETosis, a form of inflammatory programmed cell death, has the potential to precipitate harmful pathologies, including small vessel vasculitis, sepsis, systemic lupus erythematosus, and deep vein thrombosis, illustrating its significant yet potentially detrimental role in these conditions 257 , 268 , 269 ( Fig. 7 ) .

Novel HPTMs: dual roles in cardiac repair and fibrosis. When MI occurs, monocytes undergo metabolic reprogramming, leading to dysregulated glycolysis and consequently increased lactate production. The generated lactate is transported into monocytes via MCT1 and accumulates intracellularly, providing the substrate for histone la. Upon histone la, specific reparative genes such as Lrg1, Vegf-a, and IL-10 are activated and expressed. The expression of these genes modulates the dual anti-inflammatory and pro-angiogenic activities of monocytes, ultimately promoting cardiac repair. In additional, the activation of PAD4 is triggered by an increase in calcium ion concentration, leading to histone cit and the formation of NETs. NETs are composed of DNA, histones, and antimicrobial proteins and are released into the extracellular space. These NETs promote inflammatory responses, damage cardiomyocytes and endothelial cells, and interact with platelets, promoting the release of TGFβ, which in turn promotes myofibroblast fibrosis. NET formation and fibrosis are increased, whereas mice lacking PAD4 exhibit reduced fibrosis and maintain good cardiac function
Metabolic disease
An increasing body of research suggests that epigenetic mechanisms mediated by histone modifications may constitute a potential etiology for Type 2 Diabetes. In a prediabetic state, researchers conducted a systematic analysis of HPTMs of histones in the liver of a mouse model of obesity induced by a high-fat diet. They uncovered novel alterations in modifications such as ac, bu, ma, and succ, hinting that these recently revealed histone ac events may play a latent role in the development and progression of diabetes and obesity. Notably, the study also found that metformin could reverse certain specific histone modification markers, such as changes in histone H3K36ma2. 15 Research indicates that levels of malonyl-CoA are elevated in the muscles of obese and type 2 diabetes patients, underscoring the possibility that certain histone modifications may be intricately linked with the progression of these conditions. 270 In particular, bhb has been identified as a marker of active genes during starvation or diabetes ketosis induced by streptozotocin, with this modification closely associated with the metabolic pathways of the starvation response. Additionally, ketogenesis stimulated by starvation increased both bhb and bu, while the observed rise in malonylated proteins in a diabetes mouse model affected glucose and fatty acid metabolic pathways, suggesting that these modifications could potentially reverse insulin resistance. 17 , 271 , 272
The ablation of Pkm2 in endothelial cells leads to diminished serum lactate levels, which subsequently influences histone la within bone marrow stromal stem cells (BMSCs). This particular la modification is crucial for the differentiation of BMSCs into osteoblasts, which are the principal cells responsible for bone synthesis. Histone H3K18 la modulates several genes vital for osteogenesis, such as type I collagen α2 chain (COL1A2), cartilage oligomeric matrix protein, ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), and transcription factor 7-like 2. Therapeutic strategies such as the upregulation of PKM2 in endothelial cells, exogenous lactate supplementation, and physical exercise have shown efficacy in reversing the compromised phenotype in mice with a deficit in endothelial PKM2, indicating promising avenues for osteoporosis treatment. 273
Reproductive disease
Emergent HPTMs are associated with sperm maturation impairments and developmental anomalies that may impact male fertility. The bromodomains of BRDT can recognize histone ac and recruit transcription complexes to chromatin, thereby promoting the expression of specific genes. 274 Studies suggest that ac and bu at H4K5 and H4K8 positions compete at gene promoters bound by highly active BRDT, with bu also marking the delayed removal of histones during late spermatogenesis. 72 Research has unveiled that the chromodomain Y-like transcriptional corepressor (CDYL) acts as a negative regulator of histone cr. Functioning as a crotonyl-CoA hydratase, CDYL catalyzes the conversion of crotonyl-CoA to β-hydroxybutyryl-CoA, effectively diminishing the pool of crotonyl-CoA available for the crotonylation of histone lysine residues. The function of CDYL-mediated histone cr regulation is particularly significant in spermatogenesis. Research indicates that dysregulation of histone cr in CDYL transgenic mice leads to decreased male fertility, characterized by reduced epididymal sperm counts and impaired sperm motility. The study suggests that histone cr regulation by CDYL might be associated with spermatogenic failure in infertile males with AZFc deletions. This link may provide a new avenue for exploring therapeutic strategies or diagnosing certain types of male infertility. 114 There exists a clear necessity for additional studies to deepen our comprehension of how novel HPTMs might influence reproductive anomalies.
Neuropsychiatric disease
Epigenetic modifications have been found to be of significant importance in neurological conditions such as PD, Huntington’s disease, AD, and Major Depressive Disorder (MDD). 275 , 276 , 277 Contemporary studies propose that the onset of MDD may be intricately tied to several physiological and biochemical phenomena. These include the activation of the corticotropin-releasing hormone (CRH)/hypothalamic-pituitary-adrenal axis, excessive stimulation of the sympathetic nervous system, irregular secretion patterns of monoaminergic neurotransmitters, increased production of pro-inflammatory cytokines, diminished levels of neurotrophic factors, and significant epigenetic modifications. 278 , 279 , 280 , 281 , 282 , 283 Epigenetics may serve as one of the bridges linking environmental and genetic factors. Stressful events can lead to alterations in epigenetic modifications, thereby causing changes in gene expression.
MDD emerges as a complex condition influenced by a broad array of factors, including environmental, genetic, psychological, and biological determinants. Those afflicted with MDD may endure profound symptoms that encompass enduring and intense feelings of sadness and desolation, cognitive impairments, anhedonia, diminished verbal and motor activity, along with disruptions in sleep patterns. 284 , 285 Contemporary research indicates that significant stress events can precipitate modifications in histone configurations within the human brain, catalyzing transcriptional alterations that may culminate in the onset of MDD. Histone ma, ac, phosphorylation, cr, and bhb have all been identified as modifications intricately linked with the pathogenesis of MDD. Of particular interest in our discussion are the relationships between histone cr, bhb, and MDD. 286 The investigation conducted by Liu and colleagues illuminates that MDD, precipitated by sustained social defeat stress, correlates with a diminished presence of histone crotonylation. This reduction is observed alongside the upregulated expression of CDYL, suggesting a potential mechanistic interplay in the manifestation of MDD. This study is the first and, to date, the only one that demonstrates the link between MDD and histone crotonylation. Initially, researchers discovered that β-hydroxybutyrate, as a ketone body, plays a significant role in neuro-related disorders. It was found that bhb can protect neurons from toxic damage and prevent the degenerative changes in dopaminergic neurons seen in Alzheimer’s and Parkinson’s diseases. 146 , 147 Recent research suggests that β-hydroxybutyrate may possess antidepressant effects for Major MDD induced by chronic unpredictable stress. 287 , 288 , 289 Chen and colleagues were the first to associate the antidepressant effects of β-hydroxybutyrate with histone modifications. In mice with MDD induced by spatial restriction stress, they observed a reduction in the levels of H3K9bhb. Injections of β-hydroxybutyrate were found to increase both β-hydroxybutyrate and H3K9bhb levels, as well as enhance the expression of brain-derived neurotrophic factor (BDNF). 290 This deduction posits that H3K9bhb could serve as a pivotal regulatory factor for the expression of BDNF, hinting at the role of histone bhb as a significant avenue for unraveling the underpinning mechanisms of MDD.
At present, the pharmacological treatment of depression in clinical settings classifies antidepressants into seven primary categories: selective serotonin reuptake inhibitors (SSRIs), serotonin antagonists and reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, norepinephrine-dopamine reuptake inhibitors, tricyclic antidepressants, monoamine oxidase inhibitors, and melatonergic antidepressants. Each class operates via distinct mechanisms to modulate neurotransmitter systems and alleviate depressive symptoms. 291 These medications chiefly exert their effects by inhibiting the activity of serotonin and norepinephrine. However, approximately 40% of MDD patients exhibit insensitivity to these drugs. Consequently, the development of more effective and less toxic antidepressant medications is imperative. 292 Furthermore, the emerging focus among researchers on antidepressants that influence histone modifications, especially HDAC inhibitors, highlights a novel approach in the therapeutic landscape of MDD. These inhibitors have shown promise in alleviating symptoms of MDD by modulating epigenetic mechanisms. Bhb may be a critical factor in ketogenic diets, exhibiting broader and more pronounced neuroprotective effects in ameliorating refractory epilepsy compared to traditional restrictive diets. 293
In the study of pulmonary fibrosis, it was found that lung myofibroblasts, under the stimulation of TGF-β1, exhibit increased glycolytic activity, leading to the substantial production of lactate. This lactate accumulates not only in vitro cultures but is also detected in mice with a model of pulmonary fibrosis. Lactate promotes histone la via p300, enhancing the expression of pro-fibrotic genes and propelling the progression of lung fibrosis. Additionally, exposure to PM2.5 augments glycolytic activity, increasing the generation of lactate and other metabolic byproducts. The activation of the TGF-β/Smad2/3 and VEGFA/ERK pathways instigates the development of pulmonary fibrosis. The application of an LDHA inhibitor, specifically GNE-140, as a pre-treatment strategy, has been shown to significantly mitigate pulmonary inflammation and fibrosis triggered by PM2.5, showcasing its therapeutic promise in addressing such conditions. 294 , 295
Renal disorders can be categorized into acute kidney injury (AKI) and chronic kidney disease (CKD), with a subset of CKD potentially progressing to end-stage renal disease. Histone cr has been observed in renal tubular cells of healthy mouse and human kidney tissues. During acute AKI, levels of histone cr in renal tissues are elevated. This finding is replicated in vitro in cultured renal tubular cells exposed to the cytokine TWEAK, suggesting that TWEAK may be one of the factors influencing histone cr levels. Experiments involving the administration of crotonate to cultured renal tubular cells or kidneys found that crotonate treatment increased the expression of PGC-1α and sirtuin-3, and decreased the expression of CCL2. Systemic administration of crotonate in animal models can prevent the onset of AKI, maintain renal function, and prevent the decrease in PGC-1α and sirtuin-3 levels, as well as the increase in CCL2 expression. 14 Furthermore, analysis based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) suggests that proteins modified with hib are enriched in the IL-17 signaling pathway and in categories related to phagosomes. These pathways and categories are considered to be significantly associated with IgAN. The data imply that hib modifications may play a crucial regulatory role in the development and progression of IgAN. 296 Chen et al. measured the global levels of cr sites within the proteome of patients with CKD and those undergoing maintenance hemodialysis, finding decreased levels of cr in dialysis patients and increased levels in CKD patients. This suggests that cr may play a significant regulatory role in the transition from AKI to CKD. 297 The research conducted by Shimazu and colleagues presents groundbreaking findings on the linkage between β-hydroxybutyrate and renal ailments. It was revealed that β-hydroxybutyrate significantly shields murine kidneys from oxidative stress. This protective effect is achieved through the inhibition of HDAC activity and the augmentation of histone ac at the promoters of Foxo3a and Mt2, thereby unveiling a potential therapeutic avenue for renal diseases. 297
Research indicates that Salvianolic acid B can downregulate the expression of lactate dehydrogenase A (LDHA), thereby inhibiting histone la in macrophages, which effectively mitigates carbon tetrachloride-induced liver injury. By examining liver tissues and isolated Kupffer cells, it has been confirmed that Sal B affects the M1 polarization and histone la levels in macrophages. 298
In the progression of psoriasis, numerous studies have highlighted that elevated levels of lactate or lactate dehydrogenase are key factors. 299 , 300 Research has shown that elevating global la and H3K18la levels can increase the levels of the adiponectin ADIPOQ protein, 301 , 302 , 303 whereas transfection with si-LDHA reduces ADIPOQ protein levels. In the skin tissues of psoriasis patients, ADIPOQ levels are significantly reduced, and ADIPOQ possesses the potential for diagnosing psoriasis. Furthermore, the study uncovered that the downregulation of H3K18la levels inhibits the transcriptional activity of the ADIPOQ gene, which is a key factor contributing to the diminished ADIPOQ levels in psoriasis patients 304 (Fig. 8 ).

Impact of novel HPTMs on various disease pathogenesis. The figure represents the roles of novel HPTMs in different diseases. In MDD, for example, the expression of CDYL was increased and pr was inhibited; H3K9bhb and BDNF had an antidepressant effect. Spermatogenic failure is caused by the dysregulation of histone cr in AZFc deletions of infertile males. Under expression of Pkm2 regulates COL1A2, COMP, ENPP1, and TCF7L2, through histone la involving BMSCs osteoblast differentiation. In response to IL-17 and TNF stimulation, LDHA- and LDHB-mediated lactate production positively regulates histone la acetylation by inducing p300-dependent ADIPOQ protein and promoting the gene expression of pro-fibrotic genes, exacerbating the pathogenesis of psoriasis. In lungs, PM2.5 activates the TGF-β/Smad2/3 and VEGFA/ERK pathways, upregulating lactate-generating glycolysis in lung myofibroblasts through histone la to encode for collagen. The TWEAK cytokine induced cr expression, and cr had a significant effect on the regulation of the AKI-CKD transformation. The change of hib was a key regulatory action for IgAN
Targeted therapy of Novel Hptms
In this section, we will categorize the current inhibitors involved in HPTMs into three groups, summarizing their mechanisms of action and clinical applications. Additionally, we will specifically collate inhibitors that are still in the experimental stage (Table 2 ).
Inhibitors targeting enzymes of novel HPTMs
Sirt inhibitors.
Nicotinamide and its derivatives, such as nicotinamide riboside and nicotinamide mononucleotide, are important precursors of NAD + . 305 Nicotinamide acts as an endogenous inhibitor of SIRTs and can inhibit SIRT1 and SIRT2. 306 , 307 AK-7 is a selective SIRT2 inhibitor that has demonstrated improvements in behavioral and neuropathological phenotypes, extended lifespan, and ameliorated the neuropathology associated with HD in animal models. 308 , 309 β-naphthol inhibitors encompass a variety of SIRT inhibitors that possess a β-naphthol structure, including splitomicin, sirtinol, salermide, HR-73, and cambinol. 310 , 311 These compounds were discovered through in vitro cellular screenings; for instance, sirtinol and splitomicin are capable of inducing apoptosis and autophagy in cancer cells. A series of Sir2 inhibitors based on the indole structure were identified through large-scale fluorescent screening. These compounds, including EX-527, AC-93253, inauhzin, and Ro31-8220, predominantly inhibit SIRT1 and are associated with enhanced cell survival and p53 acetylation. 312 , 313 SIRT The SIRT rearranging ligand SirReal2 is a potent SIRT2 selective inhibitor that acts by inducing structural rearrangement and interacting with an unknown binding site, leading to increased acetylation of chromatin protein H3. 314 Discovered through phenotypic screening, tenovin-1 and its more water-soluble analog tenovin-6 primarily inhibit SIRT1 and SIRT2, decreasing tumor growth both in vitro and in vivo. 315 Additional SIRT inhibitors, including a range of compounds such as suramin, aristoforin, AGK2, and Tripos 360702, have demonstrated the ability to inhibit SIRT activity. These compounds show potential in inhibiting cancer cell growth and modulating cell signaling pathways. 316 , 317 , 318 , 319
While there have been significant advancements in the research of SIRT modulators over the past few decades, the studies have been uneven, and their clinical potential remains underexploited. Current investigations have adequately covered inhibitors of SIRT1 and SIRT2, yet research on inhibitors for SIRT3-7 is still lacking. Moreover, research on SIRT activators has primarily focused on SIRT1; there is a need for further exploration into activators and inhibitors for other SIRT family members to fully harness the therapeutic potential of SIRT molecules. 320
PAD inhibitors
PADs are a group of active enzymes that catalyze the irreversible HPTMs of arginine to citrulline. 321 , 322 Through this process, the structure and function of numerous target proteins are altered, including fibrinogen, TGF-β, nicotinamide N-methyltransferase, cytokines, and chemokines. 323 , 324 Cit serves as a biomarker for various diseases, particularly in instances of dysregulated PAD activity. 321 , 322 Five PAD isoforms, PAD1 through PAD4 and PAD6, have been identified in mammals. 325 PAD1, PAD2, and PAD4 localize to both the cytoplasm and the nucleus, where they can citrullinate histones and other chromatin-associated proteins. 326 All PAD isoforms, with the exception of PAD6, exhibit catalytic activity. 327 , 328 The PAD family plays a role in regulating multiple biological processes, including cell differentiation, apoptosis, innate immune responses, embryonic development, myelination, and gene regulation. 329
Inhibitors targeting PAD4 are delineated into two classes: irreversible inhibitors and reversible inhibitors. The irreversible category encompasses compounds such as F-amidine, o-Cl-amidine, BB-Cl-amidine, Cl-amidine, YW-356, o-F-amidine, Thr-Asp-F-amidine, and Thr-Asp-Cl-amidine. On the other hand, reversible inhibitors comprise agents like GSK199, streptonigrin, GSK484, along with select antirheumatic medications. These distinct groups of PAD4 inhibitors offer various therapeutic approaches for conditions involving this enzyme. 259 The mechanism by which 2-fluoroacetamidine, Cl-amidine, and F-amidine exert their inhibitory effects involves a primary assault on the carbonyl carbon of the thiolate anion at Cys645. This action precipitates the creation of a protonated tetrahedral intermediate, noted for its stability, which underscores the inhibitory process of these compounds. 330 The potency of o-F-amidine is 65 times that of F-amidine, with selectivity for PAD1 being at least sixfold higher than for PADs 2-4. 331 This indicates that o-F-amidine has significant advantages in selectivity and potency. Despite the identification of numerous PAD inhibitors, the efficacy of most is limited. 332 Variations exist among covalent PAD inhibitors in terms of in vivo stability, bioavailability, and isozyme selectivity, such as D-Cl-amidine (a selective inhibitor for PAD1), Cl4-amidine (selective for PAD3), and Thr-Asp-F-amidine (TDFA, a selective inhibitor for PAD4). 321 Most reversible inhibitors, like minocycline, paclitaxel, and streptonigrin, are weaker PAD inhibitors, yet GSK484 and GSK199 are potent and selective inhibitors targeting PAD4. 333 Cl-amidine, a haloacetamidine class PAD inhibitor, exhibits a higher selectivity for PAD4. It can prevent vascular abnormalities, arterial thrombosis, endothelial dysfunction, and aberrant vascular repair in systemic lupus erythematosus, as well as inhibit the formation of NETs, reduce the area of atherosclerotic lesions, and prolong carotid thrombus formation time in Apolipoprotein E knockout mice. 334 Recent studies have demonstrated that PAD4 inactivation can protect murine hearts from damage after myocardial infarction/reperfusion injury. 265 Furthermore, the Cl-amidine analog YW3-56, with improved bioavailability, can modify the gene encoding the upstream inhibitor of the mammalian target of rapamycin complex 1, SESN2. 335 Treatment with Cl-amidine reduces histone citrullination in neutrophils and prevents NET formation. 336 BB-Cl-amidine, a second-generation PAD inhibitor, alters T-cell immune responses and decreases the severity of inflammation in arthritis. 337 Both Cl-amidine and BB-Cl-amidine are potent PAD inhibitors in various anti-tumor models. 336 , 338
The development and application of PAD inhibitors represent a significant advancement in the field of cancer therapy, targeting the cit process, which is crucial in various cancer-related processes including tumor growth and metastasis. 91 , 339 PAD inhibitors can significantly decrease the proliferation of cancer cells without affecting the viability of normal cells. 340 Compounds such as Cl-amidine and F-amidine have demonstrated efficacy in inducing differentiation and apoptosis in various cancer cell lines, including HL60, HT29, TK6, and U2OS. 341 PAD4 inhibitors have been used clinically to prevent tumor dissemination and treat cancer-associated thrombosis. Effective PAD inhibitors can enhance anti-tumor activity by inhibiting CitH3 in tumors; for instance, in castration-resistant prostate cancer, PAD2-H3Cit26 is considered a novel therapeutic target. 223 , 342 Moreover, the combination of PAD inhibitors and HDAC inhibitors is regarded as a strategy for cancer therapy. Compounds such as paclitaxel, minocycline, and streptomycin have been identified as PAD inhibitors; however, due to their poor binding efficacy, they exhibit relatively weak and suboptimal therapeutic effects. 343 Newer compounds such as O-F-amidine and O-Cl-amidine have shown greater therapeutic efficacy, selectivity, and bioavailability. O-F-amidine, in particular, is markedly more effective than its predecessors, demonstrating an activity 65 times stronger than that of F-amidine and exhibiting a greater preference for PAD1 inhibition. 344 Recent developments include YW3-56, which not only inhibits PAD4 but also modulates key signaling pathways such as mTORC1, and can obstruct the autophagy of cancer cells, thereby inhibiting their growth. 345 Continued pharmacological innovation is needed to develop PAD inhibitors with improved selectivity, efficacy, and fewer side effects. This includes targeting specific PAD enzymes that are predominantly expressed in malignant cells without affecting normal tissues.
Histone cit has been identified as a biomarker and therapeutic target in cancer, particularly as PAD4-mediated cit of histone H3 is associated with poor clinical outcomes and a high rate of short-term mortality in patients with advanced cancer, and it can also predict the risk of venous thromboembolism. 231 , 233 , 346 Studies have also indicated that PAD2-mediated cit of histone H3 at arginine 26 promotes malignant progression in multiple myeloma and prostate cancer. 223 , 347 Targeting both PAD4 and HDAC2 concurrently emerges as a promising approach in osteosarcoma therapy. This strategy capitalizes on the regulatory capability of histone citrullination to modulate the expression of the tumor suppressor gene OKL38. Such a dual inhibition mechanism suggests a significant potential for therapeutic intervention in osteosarcoma, highlighting a nuanced understanding of histone modifications in cancer treatment. 91 , 348 Furthermore, PAD4 inhibitors are utilized to prevent tumor metastasis and thrombosis associated with insulinomas and breast cancer, 349 and to inhibit hematogenous metastasis of gastric cancer by targeting citrullination-mediated NETs with herbal compounds. 350 These findings underscore the significance of developing PAD inhibitors that target specific cit sites for cancer treatment and offer new directions for future cancer diagnostics and therapeutics.
The PAD4 protein has emerged as a promising therapeutic target for cancer, offering specific targeting capabilities and a favorable in vivo safety profile against tumor cells. Recognized as an antitumor agent, phenylboronic acid (PBA) can target both primary and metastatic tumors. In this context, researchers have endeavored to enhance PAD4 protein inhibitors by incorporating various PBA components, culminating in the development of highly targeted PAD4 inhibitors. Various experiments have revealed that the m-PBA-modified PAD4 inhibitor, labeled as 5i, exhibited pronounced antitumor activity. Importantly, compound 5i did not act by directly destroying cancer cells; instead, it played a substantial role in curbing the spread of tumor cells. Moreover, the compound named ta3 was found to reduce H3cit in the cell nucleus. The study determined that inhibitors of PBA-PAD4 effectively hinder both the proliferation and spread of breast cancer cells and significantly diminish the development of NETs within the cancerous tissue. This inhibitory action stems from the specific targeting of the PAD4 protein in the nuclei of neutrophils. Furthermore, the PBA-PAD4 inhibitor demonstrated remarkable antitumor activity, suggesting a novel approach for designing efficacious PAD4 inhibitors. 351
Evodia alkaloid effectively inhibits the la of histones and the expression of HIF1A in PCa cells, thereby obstructing the process of lactate-induced angiogenesis. Simultaneously, it enhances the transcription of Sema3A and suppresses the transcription of PD-L1, collaboratively impeding the formation of blood vessels in PCa and tumor growth. 352 Wang and colleagues demonstrated that the concurrent use of the PADI inhibitor Cl-Amidine and the AR signaling inhibitor enzalutamide leads to a combined effect, significantly suppressing the proliferation of CRPC cells in vitro and diminishing tumor growth in vivo models. Results from various studies highlight the importance of PADI2 in regulating AR during the advancement of prostate cancer, especially in CRPC, pointing to PADI as a promising therapeutic target for this condition. 223 I-BET762, I-BET726, and CPI-203 influence histone cr through the modulation of BRD4 levels, subsequently leading to the suppression of growth, movement and invasive behavior in PCa cell lines. 225
It has been demonstrated in studies that neutrophils can induce the cit of histone H3, leading to NET formation in both mouse and human multiple myeloma cells. One research has confirmed that targeting PAD4 with a novel and specific small molecule inhibitor, BMS-P5, can delay the onset of symptoms in mice with multiple myeloma and significantly inhibit tumor progression. 353
In studies of multiple sclerosis, abnormal elevation of PAD activity has been observed. To inhibit PAD, researchers developed a non-covalent inhibitor based on an α-amino acid and ethyl isocyanate core structure, where compound 23, containing an imidazole heterocycle, exhibited high selectivity and significant potency against PAD2. In animal models, compound 23 effectively reversed the physical disability induced by experimental autoimmune encephalomyelitis and reduced T-cell infiltration in the brain. This suggests that compound 23 and its analogs hold promise for further development as potential therapeutics for the treatment of multiple sclerosis. 354
Research indicates the roles of NETs and PAD in a model of endotoxin shock. The study utilized the PAD inhibitor YW3-56, which was found to effectively prolong the survival of mice induced with lipopolysaccharide. The efficacy of the therapeutic intervention on NETs, pro-inflammatory cytokines (including IL-6, TNFα, and IL-1β), and pulmonary damage was evaluated through ELISA and immunostaining methods. Findings from the study revealed that YW3-56 attenuated the inflammatory injury instigated by LPS. This attenuation was marked by the suppression of NET formation, a decrease in inflammatory cytokine levels, and a reduction in lung tissue injury, indicating its potential as a therapeutic agent. This suggests that inhibiting NET formation may serve as a novel strategy in treating endotoxin shock and related inflammation. 355
HDAC inhibitors
HDAC inhibitors are classified into four primary structural groups: hydroxamic acids, cyclic peptides, short-chain fatty acids, and benzamides. This categorization is predicated on the distinctive chemical structures that dictate the inhibitors’ interaction with HDAC enzymes, guiding their use and specificity in epigenetic therapies. 356 , 357 The impact of these HDAC inhibitors on HDACs and their encoding genes has been investigated in animal models of MDD, indicating their antidepressant effects. Trichostatin A (TSA) is capable of reversing hippocampal transcriptome alterations in rats induced by maternal care during early life. 358 Vorinostat, recognized in the scientific community as SAHA, holds the distinction of being the inaugural HDAC inhibitor sanctioned by the U.S. Food and Drug Administration for clinical application. Research into Vorinostat has unveiled its capability to ameliorate behaviors associated with MDD and to restore the expression of Glial Cell Line-Derived Neurotrophic Factor (GDNF) in mice subjected to Chronic Unpredictable Mild Stress, underscoring its potential for therapeutic intervention in neuropsychiatric disorders. 359 , 360 Valproic acid (VPA) has been found to influence the expression of BDNF, GSK-3β, CORT, and MC4R, and exhibits antidepressant properties. 361 , 362 , 363 MS-275, as a selective inhibitor targeting Class I HDACs, affects the expression of CREB, BDNF, CORT, RAC1 and GJA5. 364 Although HDAC inhibitors have demonstrated antidepressant properties in animal models, there remain limitations to be addressed before their widespread clinical application, including the potential inhibitory effects on the ac of non-histone proteins such as alpha-tubulin, HIF-1 alpha, Stat3, and beta-catenin. 365 , 366 , 367 , 368 For instance, Farydak, an HDAC inhibitor approved by the FDA for the treatment of multiple myeloma, has exhibited severe side effects such as gastrointestinal toxicity, thrombocytopenia, bone marrow suppression, fatal cardiac ischemic events, arrhythmias, electrocardiogram alterations, local and systemic infections, as well as hepatic dysfunction. 369 Studies have shown that combining HDAC inhibitors with common antidepressants such as fluoxetine can significantly reduce MDD-related behaviors, suggesting that HDAC inhibitors may have potential for use in conjunction with conventional antidepressants in the future treatment of MDD. Lithium has been found to decrease the expression of HDAC1, 3, 4, 5, 7, 8, and 10. 370 , 371 Olanzapine enhanced the ac of histone H3 at the promoter regions of BDNF in the hippocampal area of rats with MDD, while concurrently inhibiting HDAC5. 372 Combined treatment with lithium and valproic acid has been shown to induce BDNF expression and exhibit neuroprotective effects in MDD. 373 , 374
Effective compounds found in Xiao Yao San, such as quercetin, rutin, saikosaponin D, ferulic acid, and curcumin, have been identified through high-performance liquid chromatography (HPLC). 375 These compounds have shown potential in modulating histone modifications and treating depression, as seen in quercetin’s ability to regulate HDAC and HAT activities, ameliorate cognitive deficits, and enhance the expression of neural plasticity markers. 376 The realm of histone modification extends beyond the treatment of MDD and is being explored as a therapeutic target for an array of neurological conditions such as PD, AD, HD, and SMA. For instance, HDAC inhibitors like TSA, SAHA, VPA, and sodium butyrate have demonstrated efficacy in rodent models of Parkinson’s Disease. These inhibitors elevate the expression of neurotrophic factors, including GDNF and BDNF, safeguard dopaminergic neurons, and enhance dopamine synthesis, thereby manifesting their potential in neuroprotective strategies. 377 , 378 , 379 , 380 The neurotoxicity of α-synuclein can also be mitigated by HDAC inhibitors, thereby ameliorating the symptoms of Parkinson’s Disease. 381 Levodopa, a medication commonly used for PD, has been found to reduce the ac of histone H4. 382 VPA can inhibit beta-amyloid, a peptide highly associated with AD. 383 , 384 HDAC inhibitors such as tubastatin A and ACY-1215 have been found to reduce the hyperphosphorylation of tau protein in AD. 385 Donepezil, a common medication used in the treatment of AD, has been discovered to inhibit the binding between HDAC6 and the BDNF promoter in the cortex, thereby resulting in the overexpression of BDNF. 386 HDAC inhibitors, including TSA, SAHA, sodium butyrate, RGFP966, and LBH589, have been shown to ameliorate symptoms in mouse models of HD. 387 , 388 , 389 , 390 In models of Spinal SMA, a variety of HDAC inhibitors have been found to induce the expression of the survival motor neuron 1 gene, which is crucial for the disease. 391 , 392 , 393 , 394 , 395 , 396
Although HDAC inhibitors have demonstrated neuroprotective effects in animal models, their clinical application still faces challenges, such as their potential for severe side effects and the ability to penetrate the blood-brain barrier. HDAC inhibitors have been approved by the United States FDA for clinical trials in cancer treatment, but human clinical trials for depression have not yet been conducted.
Therapeutic strategies for lung cancer are closely linked with changes in histone la and cr. This finding suggests a potential new approach to improve PEM sensitivity. Proteomic studies of histones have uncovered the pivotal role of the HDAC1 in deacetylation complex in the deacetylation of lysine cr at the H3K18cr site on histone 3. One research has notably observed that HDAC1 inhibition diminishes both the presence of H3K18cr histone and RNA polymerase II at the caspase-1 promoter in cellular environments. Moreover, reducing HDAC1 levels is found to significantly decrease the growth rate of NSCLC cells resistant to PEM. These insights suggest that HDAC1, when crotonylated and associated with caspase-1, could play a role in influencing PEM resistance by specifically acting on GSDMD. 397
Glioma histone deacetylase inhibitors are recognized for their anti-tumor properties. These elements have been noted to initiate multiple types of histone post-translational modifications in glioma cells, notably including acetylation and bu. This suggests a profound connection between their antitumor efficacy and the processes of histone acetylation and bu. Assessing the levels of histone bu and pr in cancer cells can provide indirect insights into the pharmacological actions of HDAC inhibitors. 398
Studies have indicated that the HDAC inhibitor, SAHA, originally used for treating cutaneous T-cell lymphoma, also shows substantial effectiveness against neuroblastoma. Through detailed quantitative proteomic assessments, a significant induction of histone bu and acetylation was observed following SAHA treatment. This highlights the crucial role of HPTMs in its anti-tumor activity and encourages the exploration of novel anti-tumor drugs that target histone bu as a primary mechanism. 399
The findings of the study suggest that the primary mechanism of the anti-cancer properties of various drugs lies in their ability to target histone succ levels and the enzymes associated with the succ process. For example, the ability of aspirin to limit tumor proliferation is associated with its capacity to reduce the succ level of PGAM1. This action effectively impedes the proliferation of liver cancer cells and disrupts the glycolytic pathway. 188 One research indicates that suppressing the expression of succinyltransferases, such as KAT2A, reduces tumor cell proliferation and overall tumor growth. 126 This suggests a promising approach for utilizing histone succ in tumor therapies. Furthermore, it has been observed that aspirin inhibits glycolysis and increases the efficacy in cancer cells by reducing the total bib of ENO1. 400
Resistance to temozolomide (TMZ) remains a significant obstacle in the treatment of GBM. 401 The clinically approved antiepileptic drug stiripentol can cross the blood-brain barrier and inhibit the activity of lactate dehydrogenase A/B (LDHA/B), acting as a la inhibitor and rendering GBM cells more sensitive to TMZ both in vitro and in vivo. 179 Furthermore, an increase in mitochondrial reactive oxygen species (mROS) and glycolysis has been identified in pulmonary hypertension. 402 Investigations have uncovered that hypoxia-driven mitochondrial mROS hinder the hydroxylation process of HIF-1α. This obstruction fosters a glycolytic shift within Pulmonary Artery Smooth Muscle Cells (PASMCs) by the activated HIF-1α/PDK1&PDK2/phosphorylated-PDH-E1α pathway, leading to an escalated build-up of lactate and histone la. Such an increase in histone la at loci of HIF-1α target genes, which include Bmp5, Trpc5, and Kit, is linked to the promotion of PASMC proliferation. Diminishing Pdk1&2 levels tempers lactate concentration, histone la markers, and the proliferation of PASMCs. Additionally, pharmacological intervention with lactate dehydrogenase inhibitors has been shown to curtail histone la and mitigate PASMC proliferation and vascular remodeling in rats with hypoxic Pulmonary Hypertension. 403 The combined use of histone la and macroautophagy/autophagy inhibitors with bevacizumab therapy has demonstrated significant therapeutic efficacy in preclinical models of patients resistant to bevacizumab. 404
Inhibitors indirectly interfering with the process of novel HPTMs
Research indicates that the expression of hexokinase 2 (HK2) plays a pivotal role in activating hepatic stellate cells through lactylation-mediated histone regulation of gene expression. Targeting HK2 can inhibit the activation of hematopoietic stem cells and reduce liver fibrosis systemically, showcasing the potential of HK2 as an effective therapeutic target for liver fibrosis. 405
Research reveals that lactate influences the treatment and prognosis of CRC by inhibiting RARγ, primarily by remodeling the functionality of macrophages within the tumor microenvironment. Additionally, the study identifies nordihydroguaiaretic acid (NDGA) as an effective therapeutic agent that directly binds to RARγ and inhibits the TRAF6-IL-6-STAT3 signaling pathway, offering a new therapeutic strategy targeting pro-tumoral macrophages in CRC. 406 In another study, researchers conducted a global mapping of HPTMs in CRC cells treated with largazole-7. They noted that the drug’s selectivity for cancer cells was more than 100 times greater than that for normal cells. Changes in lysine methylation and bu were observed at 68 core histone sites following drug exposure, suggesting that largazole-7 could counteract lysine bu. 407 This finding suggests that the anti-tumor efficacy and high specificity of largazole-7 may be attributed to its targeted action against histone bu in CRC cells. Additionally, it has been demonstrated that the cancer treatment delivery ligand, GnRH-III, shows enhanced tumor-inhibitory effects and higher binding affinity when combined with bu. 408 , 409 Moreover, precise rectification of irregular histone la markedly restrains both the development and spread of ccRCC in live models. Even more crucial is the finding that simultaneously targeting histone la and PDGFRβ greatly amplifies the treatment effectiveness. This research underscores the vital importance of HPTMs, particularly histone la, in the advancement of ccRCC, indicating that interrupting the reinforcing cycle between histone la and PDGFRβ signaling may provide an innovative approach to treating ccRCC patients. 209
Pan et al. identified a unique triterpene anti-tumor compound, DML and demonstrated its efficacy in inhibiting HCC progression by targeting two tumor-promoting HPTMs sites: H3K9la and H3K56la. The in vivo mechanism of DML in regulating H3 la was confirmed through a tumor xenograft model in nude mice, highlighting DML’s potential as a promising candidate for HCC treatment. 410 Additionally, A link between la of H3 histone and the anticancer properties of RJA has been demonstrated in studies. Their research revealed that RJA impedes the progression of HCC by disrupting lactate generation and blocking la at the histone sites H3K9la and H3K14la. 411
It has been discovered that glioblastoma stem cells contribute to the accumulation of crotonyl-CoA and the cr of histone H4 lysine by regulating the mechanism of lysine catabolism. Furthermore, experimental evidence has shown that inhibiting histone lysine cr can effectively suppress tumor progression. 412
Andrographolide (AGP) can significantly reduce aortic valve calcification by inhibiting the p300 enzyme’s la of histones, particularly at the H3K9 and H3K18 sites. This la is associated with the expression of Runx2, a key factor influencing bone metabolism and calcification. By inhibiting these la sites, AGP diminishes Runx2 expression, thereby mitigating calcification. 413
Common techniques and recent advances in Hptms research
In the domain of epigenetics research, a plethora of sophisticated technologies have been developed to study the epigenomic states of genomes and their underlying molecular mechanisms. ChIP-seq is utilized to investigate protein-DNA interactions and histone modifications across entire genomes. 414 , 415 DNase-seq and ATAC-seq assess chromatin accessibility, with the latter being particularly suited for samples with a low cell count. 416 , 417 FAIRE-Seq 418 and MNase-seq 419 are employed to identify open chromatin regions and map nucleosome positions. High-throughput sequencing strategies such as BS-Seq, 420 oxBS-Seq, 421 fCAB-Seq, 422 and CAB-Seq 423 have been formulated to explore DNA and RNA modifications. Novel techniques like CUT&Tag and CUT&RUN leverage antibodies to provide high-resolution and cost-effective solutions for analyzing transcription factors, histone modifications, and protein-DNA interactions. 424 , 425 , 426 Each technique comes with its own set of strengths and limitations, and often a combination of multiple methods is required. With growing demands, it becomes especially critical to develop more streamlined and practical technologies for the future ( Fig. 9 ) .

Timeline of HPTMs research techniques. a The progression in researchers’ methods for studying histones from the early 20th century to the early 21st century. b The specific advancements and technical developments in studying histones based on NGS, TGS, and MS over the past 15 years
Next-generation sequencing technology (NGS)
NGS technologies has greatly expanded the methodologies available for protein research, particularly through the integration of ChIP with sequencing known as ChIP-Seq, enabling scientists to comprehensively map epigenetic marks across the entire genome. 427 , 428 This is particularly significant for the prognosis of diseases and the development of therapies. Since its first application by Barski and colleagues in 2007, ChIP-Seq has become a pivotal tool for studying epigenetic modifications due to its cost-effectiveness, efficiency, high sensitivity, and extensive genomic coverage. 429
In 2017, the Henikoff laboratory introduced CUT&RUN, a technique that addresses issues of high false-positive rates and poor antibody specificity seen in traditional ChIP-seq. Its advantages include applicability to small cell samples, high signal-to-noise ratio, low cell requirements, and the ability to detect distal transcription factor binding within three-dimensional space. 426 , 430 The CUT&Tag technology, introduced in 2019, precisely identifies and characterizes characteristic peaks of histone modifications through methods such as GoPeaks, suitable for genome-wide analysis, and can be combined with single-cell library construction techniques for high-resolution measurements, yielding high-quality single-cell chromatin modification data. 425 , 431 The CUT&Tag-BS method enables simultaneous detection of histone modifications and DNA methylation, particularly fitting for limited sample volumes. 432
Advancements in single-cell sequencing technologies have led to the development of ACT-seq, which allows for the analysis of histone tail modifications in minute quantities, including single cells, and enables the construction of thousands of single-cell libraries within a single day. 433 Moreover, Yang and colleagues have developed the scChIX-seq technique, which combines experimental and computational methods to concurrently analyze multiple histone marks within individual cells. This approach supports multimodal analysis of antibody-mediated chromatin studies, endowing scChIX-seq with extensive potential applications in epigenetic research 434 (Table 3 ).
Third-generation sequencing (TGS)
TGS, also known as single-molecule sequencing, operates by allowing a single DNA strand to pass through a nanopore, with the sequence of bases identified using fluorescence or electrical blockade. TGS addresses some limitations of second-generation sequencing, particularly overcoming issues of limited read length in NGS, by enabling continuous sequencing of hundreds of thousands of bases. TGS is versatile, applicable not only to DNA and RNA sequencing but also to direct observation of epigenetic patterns on proteins and genetic material. 435 Furthermore, TGS offers distinct advantages over NGS in handling complex DNA scenarios, thus facilitating more in-depth research into histone epigenetic modifications. 436 For instance, TGS has been identified as a unique tool for investigating HPV sequences. 437
In 2014, Oxford Nanopore Technologies introduced the MiniON, the first commercially available nanopore sequencer. 438 Despite challenges like limited continuity and higher error rates in base calling, nanopore technology’s potential has been widely recognized. 439 The main TGS platforms are Pacific Biosciences and Oxford Nanopore Technologies, each offering unique solutions. Nanopore technology pairs each nanopore with a nucleic acid cleavage enzyme, whereas Pacific Biosciences achieves sequencing by introducing fluorescently labeled bases alongside the target sequence, using DNA polymerase. The introduction of these platforms has significantly propelled the advancement of TGS technology. 440 , 441
Moreover, novel methodologies based on nanopore technology such as nanoHiMe-seq 442 and SMOOTH-seq 443 have been developed, which can be utilized to explore the intricate interactions of epigenetic modifications within the genome or to identify structural variants and extrachromosomal circular DNA within single cells. The base accuracy rate of Third-Generation Sequencing is currently around 90%, and it also confronts the challenge of extracting large macromolecules, high-molecular-weight DNA, and intact RNA from clinical samples. 444
Mass spectrometry (MS)
MS is a precise analytical tool that measures the mass-to-charge ratio (m/z) of ionized molecules to accurately determine their molecular weight. 445 , 446 MS has emerged as the method of choice for the identification and quantification of HPTMs due to its objectivity, comprehensiveness, and precise quantification. 447 Compared to traditional antibody-based methods, mass spectrometry boasts the advantage of detecting any type of HPTM in a single experiment without prior knowledge of the modification type or location. Additionally, mass spectrometry can accurately quantify HPTMs, addressing issues such as cross-reactivity and epitope masking associated with antibody methods. 448
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) has become an integral advancement in MS-based targeted proteomics, offering high sensitivity and precision in protein analysis, and excelling particularly in the large-scale quantitative analysis of proteins and their post-translational modifications. These technologies play a crucial role in identifying and validating cancer biomarkers, facilitating early diagnosis, unraveling molecular mechanisms, and guiding therapeutic strategies. 449 , 450 , 451 Through LC/MS, researchers can analyze histone HPTM patterns in normal and tumor tissues, such as the observed decrease in H3K27me3 and an increase in H3K9me and H3K36me1/me2 in aggressive triple-negative breast cancer. 452 , 453
Despite its widespread application, LC/MS’s limitation lies in the loss of spatial information. In contrast, Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging (MALDI-MS Imaging) showcases unique advantages in the analysis of histone variants and HPTMs, especially in providing spatial information. 454 By directly applying MALDI matrix onto tissue samples and using laser scanning, this technique generates a mass spectrum for each measurement point, offering spatial distribution information akin to immunohistochemistry while simultaneously analyzing multiple peptides. MALDI Imaging has been successfully applied to identify regulated histone HPTMs/variants in various disease states, such as the increase of H4K16 acetylation and K20 dimethylation in hepatocellular carcinoma. 455 , 456 To overcome challenges associated with MALDI Imaging, such as analysis resolution and protein quantification issues, this technique is often coupled with LC/MS, providing new avenues for histone-based disease research and the identification of epigenetic markers.
While bottom-up mass spectrometry methods are popular for their high throughput and efficiency, they often sacrifice information pertaining to combinatorial HPTMs patterns and variant differences. In contrast, middle-down and top-down approaches can provide richer information but face challenges in the clinical application such as data analysis complexity and interpretation. These methods require specific instrument setups, are analytically complex, and demand larger amounts of starting material, which limits their widespread application in clinical settings. 456 Recently introduced direct infusion mass spectrometry techniques, which forgo traditional HPLC separation, can analyze 200 HPTMs within one minute, addressing reproducibility issues associated with nanoHPLC separations and demonstrating the potential to process up to 1000 samples per day. 457 This offers hope for their application in clinical samples, though the suitability of these technologies will require further validation. However, their integration with targeted MRM acquisition workflows promises to be an ideal choice for clinical sample processing. 458
Additionally, an array of pre-mass spectrometry techniques such as Trapped Ion Mobility Spectrometry (TIMS), 459 Ion Mobility Spectrometry (IMS), 460 and various fragmentation methods (such as CID, beam-type CID, ETD, and UVPD 461 ) have significantly advanced the study of HPTMs. These technologies, by enhancing the selectivity and sensitivity of analysis, have bolstered the capacity to identify and quantify protein modifications in complex biological samples. Advances in high-resolution mass spectrometry technologies, such as Orbitrap 462 and FTICR, 463 are crucial for identifying subtle mass differences in molecules, especially in the study of HPTMs. These technologies provide precise mass measurements, aiding in a deeper understanding of the roles proteins play in cellular functions and disease progression.
BioID technology is an effective and widely used method for proximity-dependent labeling of proteins in eukaryotic cells. This technique is designed to screen for interacting and proximal proteins within their natural cellular environment, and it has also been utilized in histone research. 464 Additionally, technologies like BioID can complement traditional affinity purification mass spectrometry (AP-MS) methods. 465 Recently, the development of in vitro BioID (ivBioID) has enabled scientists to depict the microenvironment of the histone H3 variant CENP-A with higher temporal and spatial resolution. 466 The future joint application of ivBioID and MS is also worth looking forward to. This advancement provides new perspectives and tools for studying protein interactions and cellular functions.
Conclusion and perspective
In summary, this review encapsulates the historical progress of histone modifications, their basic structures, enzymes involved, and biological functions, while also emphasizing the significant contributions of nine novel histone modifications in advancing the treatment of various diseases, particularly cancer. The review emphasizes the impact of novel histone modifications on diseases treatment, particularly through metabolic pathways, their potential in overcoming drug resistance, and their synergistic effects with other treatments. Furthermore, we also discuss the importance of advanced techniques in histone research, which are crucial for comprehending the cancer physiology. The summary of the relationship between histone modifications and diseases has enriched our understanding of diseases and led to the development of targeted inhibitors. These advancements not only hold promise for refining current diseases therapeutic strategies but also open avenues for more personalized and effective approaches to diseases management in the foreseeable future.
Although numerous researches have highlighted the importance and therapeutic potential of HPTMs in various diseases, the specific roles and mechanisms of HPTMs in the development and treatment of cancers remain largely unclear. Furthermore, the landscape of epigenetic regulation becomes even more complex when considering competitive or antagonistic interactions that may arise as different modification pathways converge on the same amino acid residue. 4 For instance, there is a balance between the ac and ma of histone H3 at lysine 9, 467 as well as a dynamic competition between ac and bu at lysines 5 and 8 of histone H4. 468 Additionally, within the field of epigenetics in cancer therapy, the roles of “writers”, “erasers” and “readers” present challenges, especially in terms of their specificity to individual HPTMs. These proteins exhibit affinity for a broad range of HPTMs, which could lead to a variety of side effects. The interaction between histone modifications and other forms of epigenetic alterations, like DNA ma, requires additional exploration. 469
Over the past few decades, significant progress has been made in the research of various histone inhibitors. However, the research remains unbalanced, and the clinical potential has not been fully exploited. Developing highly efficient, selective inhibitors of histone modifications is a vital need for future research. Dual inhibitors, such as tyrosine kinase and HDAC inhibitors, 470 , 471 VEGFR-2/HDAC inhibitors, 472 and FGFR/HDAC inhibitors, 473 , 474 hold immense application potential in cancer therapy. Moreover, several novel dual inhibitors have entered clinical trial phases, demonstrating feasibility and efficacy in the treatment of solid tumors. 475 The roles of novel HPTMs in other diseases remain worthy of investigation. For instance, emerging HPTMs continue to have a distinctive impact on processes like embryonic development; however, research into the connection between histone modifications and their developmental implications or therapeutic studies remains scant. Moreover, many traditional HPTMs are crucial regulatory factors of cellular functions. The interplay or competitive mechanisms between them and novel HPTMs and their roles in diseases require further investigation. In addition, research on the “readers” of various novel HPTMs remains a blank slate. Given the ubiquity of various acylation modifications, there must exist more types and functions of “writers,” “erasers,” and “readers” in novel HPTMs, represented by la, thus warranting further exploration. This will provide new ideas and targets for improving cancer treatment outcomes and tumor prognosis. There is an anticipation that more research in the future will delve into these diseases, providing a deeper understanding of these epigenetic influences. With the continuous advancement of novel analytical techniques, it is anticipated that more types of histone modifications and new target inhibitors will be discovered, as well as hybrids that not only possess multi-target effects but also significantly enhance therapeutic efficacy in vivo. This holds tremendous potential for the future treatment of various diseases, particularly cancer.
Huang, H. et al. SnapShot: Histone modifications. Cell 159 , 458–458.e451 (2014).
Article CAS PubMed PubMed Central Google Scholar
Recillas-Targa, F. Cancer epigenetics: An overview. Arch. Med. Res . 53 , 732–740 (2022).
Article CAS PubMed Google Scholar
Hanover, J. A., Krause, M. W. & Love, D. C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 13 , 312–321 (2012).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21 , 381–395 (2011).
Kouzarides, T. Chromatin modifications and their function. Cell 128 , 693–705 (2007).
Chen, Y. et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell Proteom. 6 , 812–819 (2007).
Article CAS Google Scholar
Tan, M. et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146 , 1016–1028 (2011).
Dai, L. et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 10 , 365–370 (2014).
Xie, Z. et al. Lysine succinylation and lysine malonylation in histones. Mol. Cell Proteom. 11 , 100–107 (2012).
Rousseaux, S. & Khochbin, S. Histone acylation beyond acetylation: Terra incognita in chromatin biology. Cell J. 17 , 1–6 (2015).
PubMed PubMed Central Google Scholar
Benayoun, B. A. et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29 , 697–709 (2019).
Zhang, K. et al. Comparative analysis of histone H3 and H4 post-translational modifications of esophageal squamous cell carcinoma with different invasive capabilities. J. Proteom. 112 , 180–189 (2015).
Shi, Y. et al. Epigenetic regulation in cardiovascular disease: mechanisms and advances in clinical trials. Signal Transduct. Target Ther. 7 , 200 (2022).
Ruiz-Andres, O. et al. Histone lysine crotonylation during acute kidney injury in mice. Dis. Model Mech. 9 , 633–645 (2016).
Nie, L. et al. The landscape of histone modifications in a high-fat diet-induced obese (DIO) mouse model. Mol. Cell Proteom. 16 , 1324–1334 (2017).
Liu, Y. et al. Chromodomain Y-like protein-mediated histone crotonylation regulates stress-induced depressive behaviors. Biol. Psychiatry 85 , 635–649 (2019).
Du, Y. et al. Lysine malonylation is elevated in type 2 diabetic mouse models and enriched in metabolic associated proteins. Mol. Cell Proteom. 14 , 227–236 (2015).
Zhao, A. et al. Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic malignancies. Signal Transduct. Target Ther. 8 , 71 (2023).
Article PubMed PubMed Central Google Scholar
Wang, N., Ma, T. & Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct. Target Ther. 8 , 69 (2023).
Xu, H. et al. Function and mechanism of novel histone posttranslational modifications in health and disease. Biomed. Res Int 2021 , 6635225 (2021).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574 , 575–580 (2019).
Baumann, K. Post-translational modifications: Crotonylation versus acetylation. Nat. Rev. Mol. Cell Biol. 16 , 265 (2015).
Xie, Z. et al. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell 62 , 194–206 (2016).
Waddington, C. H. The epigenotype. 1942. Int J. Epidemiol. 41 , 10–13 (2012).
Nanney, D. L. Epigenetic control systems. Proc. Natl. Acad. Sci. USA 44 , 712–717 (1958).
Riggs, A. D. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet 14 , 9–25 (1975).
Holliday, R. Epigenetics: An overview. Dev. Genet 15 , 453–457, (1994).
Holliday, R. & Pugh, J. E. DNA modification mechanisms and gene activity during development. Science 187 , 226–232, (1975).
Bird, A. Perceptions of epigenetics. Nature 447 , 396–398 (2007).
Peixoto, P., Cartron, P. F., Serandour, A. A. & Hervouet, E. From 1957 to nowadays: A brief history of epigenetics. Int J. Mol. Sci. 21 , 7571 (2020).
Nicoglou, A. & Merlin, F. Epigenetics: A way to bridge the gap between biological fields. Stud. Hist. Philos. Biol. Biomed. Sci. 66 , 73–82 (2017).
Article PubMed Google Scholar
Wu, Y. L. et al. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal Transduct. Target Ther. 8 , 98 (2023).
Allfrey, V. G., Faulkner, R. & Mirsky, A. E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl Acad. Sci. USA 51 , 786–794 (1964).
Kornberg, R. D. Chromatin structure: A repeating unit of histones and DNA. Science 184 , 868–871, (1974).
Brannan, C. I., Dees, E. C., Ingram, R. S. & Tilghman, S. M. The product of the H19 gene may function as an RNA. Mol. Cell Biol. 10 , 28–36 (1990).
CAS PubMed PubMed Central Google Scholar
Lee, R. C., Feinbaum, R. L., Ambros, V. & The, C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75 , 843–854 (1993).
Brownell, J. E. et al. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84 , 843–851 (1996).
Taunton, J., Hassig, C. A. & Schreiber, S. L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272 , 408–411 (1996).
Luger, K. et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389 , 251–260 (1997).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406 , 593–599 (2000).
Wu, G. et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet 44 , 251–253 (2012).
Schwartzentruber, J. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482 , 226–231 (2012).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518 , 317–330 (2015).
Grunstein, M. Histones as regulators of genes. Sci. Am. 267 , 68–74b (1992).
Turner, B. M. Histone acetylation and an epigenetic code. Bioessays 22 , 836–845 (2000).
Zaib, S., Rana, N. & Khan, I. Histone modifications and their role in epigenetics of cancer. Curr. Med Chem. 29 , 2399–2411 (2022).
Shiio, Y. & Eisenman, R. N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 100 , 13225–13230, (2003).
Du, J. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334 , 806–809 (2011).
Cavalli, G. & Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 571 , 489–499 (2019).
Lennartsson, A. & Ekwall, K. Histone modification patterns and epigenetic codes. Biochim. Biophys. Acta 1790 , 863–868 (2009).
Li, X. & Li, X. D. Integrative chemical biology approaches to deciphering the histone code: A problem-driven journey. Acc. Chem. Res 54 , 3734–3747 (2021).
Cutter, A. R. & Hayes, J. J. A brief review of nucleosome structure. FEBS Lett. 589 , 2914–2922 (2015).
Joseph, F. M. & Young, N. L. Histone variant-specific post-translational modifications. Semin Cell Dev. Biol. 135 , 73–84 (2023).
Lorch, Y., Kornberg, R. D. & Maier-Davis, B. Role of the histone tails in histone octamer transfer. Nucleic Acids Res. 51 , 3671–3678 (2023).
Li, G., Levitus, M., Bustamante, C. & Widom, J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12 , 46–53 (2005).
Cosgrove, M. S., Boeke, J. D. & Wolberger, C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 11 , 1037–1043 (2004).
North, J. A. et al. Phosphorylation of histone H3(T118) alters nucleosome dynamics and remodeling. Nucleic Acids Res . 39 , 6465–6474 (2011).
Bao, X. et al. Glutarylation of histone H4 lysine 91 regulates chromatin dynamics. Mol. Cell 76 , 660–675.e669 (2019).
Zhang, L., Eugeni, E. E., Parthun, M. R. & Freitas, M. A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma 112 , 77–86 (2003).
Valls, E., Sánchez-Molina, S. & Martínez-Balbás, M. A. Role of histone modifications in marking and activating genes through mitosis. J. Biol. Chem. 280 , 42592–42600 (2005).
Xin, L. et al. Exploring cellular memory molecules marking competent and active transcriptions. BMC Mol. Biol. 8 , 31 (2007).
Ali, I., Conrad, R. J., Verdin, E. & Ott, M. Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem. Rev. 118 , 1216–1252 (2018).
Dahlin, J. L., Chen, X., Walters, M. A. & Zhang, Z. Histone-modifying enzymes, histone modifications and histone chaperones in nucleosome assembly: Lessons learned from Rtt109 histone acetyltransferases. Crit. Rev. Biochem. Mol. Biol. 50 , 31–53 (2015).
Shibata, S. Chromatin dynamics and epigenetics in skin stress adaptation. J. Dermatol Sci. 103 , 66–72 (2021).
Liu, B. et al. Identification and characterization of propionylation at histone H3 lysine 23 in mammalian cells. J. Biol. Chem. 284 , 32288–32295 (2009).
Kaczmarska, Z. et al. Structure of p300 in complex with acyl-CoA variants. Nat. Chem. Biol. 13 , 21–29 (2017).
Han, Z. et al. Revealing the protein propionylation activity of the histone acetyltransferase MOF (males absent on the first). J. Biol. Chem. 293 , 3410–3420 (2018).
Peng, C. et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell Proteom. 10 , M111.012658 (2011).
Article Google Scholar
Sabari, B. R., Zhang, D., Allis, C. D. & Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18 , 90–101 (2017).
Zhao, D. et al. YEATS domain-A histone acylation reader in health and disease. J. Mol. Biol. 429 , 1994–2002 (2017).
Bao, X. et al. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. Elife 3 , e02999 (2014).
Zhao, S., Zhang, X. & Li, H. Beyond histone acetylation-writing and erasing histone acylations. Curr. Opin. Struct. Biol. 53 , 169–177 (2018).
Xie, Y. et al. The role and mechanism of histone lactylation in health and diseases. Front Genet 13 , 949252 (2022).
Vaupel, P. & Multhoff, G. Revisiting the Warburg effect: historical dogma versus current understanding. J. Physiol. 599 , 1745–1757 (2021).
Chen, L. et al. Lactate-lactylation hands between metabolic reprogramming and immunosuppression. Int J. Mol. Sci. 23 , 11943 (2022).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324 , 1029–1033, (2009).
Liberti, M. V. & Locasale, J. W. Histone lactylation: A new role for glucose metabolism. Trends Biochem Sci. 45 , 179–182 (2020).
Cluntun, A. A. et al. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 3 , 10 (2015).
Wu, J. et al. Stromal-epithelial lactate shuttle induced by tumor-derived interleukin-1β promotes cell proliferation in oral squamous cell carcinoma. Int J. Mol. Med 41 , 687–696 (2018).
CAS PubMed Google Scholar
Doherty, J. R. & Cleveland, J. L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Invest 123 , 3685–3692 (2013).
Moreno-Yruela, C. et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci. Adv. 8 , eabi6696 (2022).
Desgeorges, T. et al. Histone lactylation in macrophages is predictive for gene expression changes during ischemia induced-muscle regeneration. Mol. Metab. 83 , 101923 (2024).
Dai, X., Lv, X., Thompson, E. W. & Ostrikov, K. K. Histone lactylation: Epigenetic mark of glycolytic switch. Trends Genet 38 , 124–127 (2022).
Rogers, G. E. & Simmonds, D. H. Content of citrulline and other amino-acids in a protein of hair follicles. Nature 182 , 186–187 (1958).
van Boekel, M. A., Vossenaar, E. R., van den Hoogen, F. H. & van Venrooij, W. J. Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res 4 , 87–93 (2002).
Kroot, E. J. et al. The prognostic value of anti-cyclic citrullinated peptide antibody in patients with recent-onset rheumatoid arthritis. Arthritis Rheum. 43 , 1831–1835 (2000).
Ishida-Yamamoto, A. et al. Decreased deiminated keratin K1 in psoriatic hyperproliferative epidermis. J. Invest Dermatol 114 , 701–705 (2000).
Chumanevich, A. A. et al. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol. 300 , G929–G938 (2011).
Ishigami, A. et al. Abnormal accumulation of citrullinated proteins catalyzed by peptidylarginine deiminase in hippocampal extracts from patients with Alzheimer’s disease. J. Neurosci. Res 80 , 120–128 (2005).
Vossenaar, E. R. et al. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann. Rheum. Dis. 63 , 373–381 (2004).
Li, P. et al. Coordination of PAD4 and HDAC2 in the regulation of p53-target gene expression. Oncogene 29 , 3153–3162 (2010).
Hagiwara, T. et al. Deimination of arginine residues in nucleophosmin/B23 and histones in HL-60 granulocytes. Biochem. Biophys. Res Commun. 290 , 979–983 (2002).
Zhang, X. et al. Peptidylarginine deiminase 1-catalyzed histone citrullination is essential for early embryo development. Sci. Rep. 6 , 38727 (2016).
Golenberg, N. et al. Citrullination regulates wound responses and tissue regeneration in zebrafish. J. Cell Biol. 219 , e201908164 (2020).
Sekeri-Pataryas, K. E. & Sourlingas, T. G. The differentiation-associated linker histone, H1.0, during the in vitro aging and senescence of human diploid fibroblasts. Ann. NY Acad. Sci. 1100 , 361–367 (2007).
Cherrington, B. D. et al. Potential role for peptidylarginine deiminase 2 (PAD2) in citrullination of canine mammary epithelial cell histones. PLoS One 5 , e11768 (2010).
Khan, S. A. et al. GnRH stimulates peptidylarginine deiminase catalyzed histone citrullination in gonadotrope cells. Mol. Endocrinol. 30 , 1081–1091 (2016).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303 , 1532–1535 (2004).
Khandpur, R. et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med 5 , 178ra140 (2013).
Li, P. et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med 207 , 1853–1862 (2010).
Wu, Z. et al. Inhibition of PAD2 improves survival in a mouse model of lethal LPS-induced endotoxic shock. Inflammation 43 , 1436–1445 (2020).
Zuo, Y. et al. Neutrophil extracellular traps in COVID-19. JCI Insight 5 , e138999 (2020).
Chang, X. et al. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 9 , 40 (2009).
Zhu, D., Zhang, Y. & Wang, S. Histone citrullination: a new target for tumors. Mol. Cancer 20 , 90 (2021).
Berger-Achituv, S. et al. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 4 , 48 (2013).
Tohme, S. et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res 76 , 1367–1380 (2016).
Demers, M. et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 5 , e1134073 (2016).
Chen, Y., Chen, W., Cobb, M. H. & Zhao, Y. PTMap–a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc. Natl. Acad. Sci. USA 106 , 761–766 (2009).
Dai, S. K. et al. Histone crotonylation regulates neural stem cell fate decisions by activating bivalent promoters. EMBO Rep. 22 , e52023 (2021).
Liu, K. et al. A qualitative proteome-wide lysine crotonylation profiling of papaya (Carica papaya L.). Sci. Rep. 8 , 8230 (2018).
Sabari, B. R. et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 69 , 533 (2018).
Liu, X. et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 3 , 17016 (2017).
Wan, J., Liu, H., Chu, J. & Zhang, H. Functions and mechanisms of lysine crotonylation. J. Cell Mol. Med . 23 , 7163–7169 (2019).
Liu, S. et al. Chromodomain protein CDYL acts as a crotonyl-CoA hydratase to regulate histone crotonylation and spermatogenesis. Mol. Cell 67 , 853–866.e855 (2017).
Fu, H. et al. Dynamics of telomere rejuvenation during chemical induction to pluripotent stem cells. Stem Cell Rep. 11 , 70–87 (2018).
Jiang, G. et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J. Clin. Invest 128 , 1190–1198 (2018).
Wan, J. et al. HOXB9 promotes endometrial cancer progression by targeting E2F3. Cell Death Dis. 9 , 509 (2018).
Kawai, Y. et al. Formation of Nepsilon-(succinyl)lysine in vivo: a novel marker for docosahexaenoic acid-derived protein modification. J. Lipid Res 47 , 1386–1398 (2006).
Zhang, Z. et al. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7 , 58–63 (2011).
Li, X. et al. Systematic identification of the lysine succinylation in the protozoan parasite Toxoplasma gondii. J. Proteome Res 13 , 6087–6095 (2014).
Tan, M. et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 19 , 605–617 (2014).
Ye, J. et al. Histone H4 lysine 91 acetylation a core domain modification associated with chromatin assembly. Mol. Cell 18 , 123–130 (2005).
Hirschey, M. D. & Zhao, Y. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol. Cell Proteom. 14 , 2308–2315, (2015).
Zorro Shahidian, L. et al. Succinylation of H3K122 destabilizes nucleosomes and enhances transcription. EMBO Rep. 22 , e51009 (2021).
Yokoyama, A., Katsura, S. & Sugawara, A. Biochemical analysis of histone succinylation. Biochem Res Int 2017 , 8529404 (2017).
Wang, Y. et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 552 , 273–277 (2017).
Simithy, J. et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 8 , 1141 (2017).
Trefely, S., Lovell, C. D., Snyder, N. W. & Wellen, K. E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 38 , 100941 (2020).
Tong, Y. et al. KAT2A succinyltransferase activity-mediated 14-3-3ζ upregulation promotes β-catenin stabilization-dependent glycolysis and proliferation of pancreatic carcinoma cells. Cancer Lett. 469 , 1–10 (2020).
Alleyn, M., Breitzig, M., Lockey, R. & Kolliputi, N. The dawn of succinylation: A posttranslational modification. Am. J. Physiol. Cell Physiol. 314 , C228–c232 (2018).
Nathan, D. et al. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 20 , 966–976 (2006).
Dhall, A. et al. Chemically sumoylated histone H4 stimulates intranucleosomal demethylation by the LSD1-CoREST complex. ACS Chem. Biol. 12 , 2275–2280 (2017).
Hsu, P. L. et al. Structural basis of H2B ubiquitination-dependent H3K4 methylation by COMPASS. Mol. Cell 76 , 712–723.e714 (2019).
Zhang, K., Chen, Y., Zhang, Z. & Zhao, Y. Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. J. Proteome Res 8 , 900–906 (2009).
Kebede, A. F. et al. Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol. 24 , 1048–1056 (2017).
Vollmuth, F. & Geyer, M. Interaction of propionylated and butyrylated histone H3 lysine marks with Brd4 bromodomains. Angew. Chem. Int Ed. Engl. 49 , 6768–6772, (2010).
Lin, H., Su, X. & He, B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem. Biol. 7 , 947–960, (2012).
Kaelin, W. G. Jr. & McKnight, S. L. Influence of metabolism on epigenetics and disease. Cell 153 , 56–69 (2013).
Wellen, K. E. et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324 , 1076–1080 (2009).
Guenzel, A. J. et al. Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol. Ther. 21 , 1316–1323 (2013).
Huang, G. et al. An information entropy-based approach for computationally identifying histone lysine butyrylation. Front Genet 10 , 1325 (2019).
Zhu, Z. et al. Identification of lysine isobutyrylation as a new histone modification mark. Nucleic Acids Res. 49 , 177–189 (2021).
Maxwell, P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399 , 271–275 (1999).
Katada, S., Imhof, A. & Sassone-Corsi, P. Connecting threads: epigenetics and metabolism. Cell 148 , 24–28, (2012).
Marosi, K. et al. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem 139 , 769–781 (2016).
Kashiwaya, Y. et al. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl Acad. Sci. USA 97 , 5440–5444 (2000).
Tieu, K. et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest 112 , 892–901 (2003).
Liu, K. et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 10 , 243 (2019).
Millán-Zambrano, G., Burton, A., Bannister, A. J. & Schneider, R. Histone post-translational modifications - cause and consequence of genome function. Nat. Rev. Genet 23 , 563–580 (2022).
Durrin, L. K., Mann, R. K., Kayne, P. S. & Grunstein, M. Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell 65 , 1023–1031, (1991).
Protacio, R. U., Li, G., Lowary, P. T. & Widom, J. Effects of histone tail domains on the rate of transcriptional elongation through a nucleosome. Mol. Cell Biol. 20 , 8866–8878, (2000).
Nitsch, S., Zorro Shahidian, L. & Schneider, R. Histone acylations and chromatin dynamics: concepts, challenges, and links to metabolism. EMBO Rep. 22 , e52774 (2021).
Sabari, B. R. et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 58 , 203–215 (2015).
Gowans, G. J. et al. Recognition of histone crotonylation by Taf14 links metabolic state to gene expression. Mol. Cell 76 , 909–921.e903 (2019).
Tidwell, T., Allfrey, V. G. & Mirsky, A. E. The methylation of histones during regeneration of the liver. J. Biol. Chem. 243 , 707–715 (1968).
Bannister, A. J., Schneider, R. & Kouzarides, T. Histone methylation: dynamic or static? Cell 109 , 801–806, (2002).
Henikoff, S. & Shilatifard, A. Histone modification: cause or cog? Trends Genet 27 , 389–396, (2011).
Talbert, P. B., Meers, M. P. & Henikoff, S. Old cogs, new tricks: the evolution of gene expression in a chromatin context. Nat. Rev. Genet 20 , 283–297 (2019).
Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131 , 58–69 (2007).
Ng, H. H., Robert, F., Young, R. A. & Struhl, K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11 , 709–719 (2003).
Petruk, S. et al. Trithorax and dCBP acting in a complex to maintain expression of a homeotic gene. Science 294 , 1331–1334 (2001).
Hörmanseder, E. et al. H3K4 methylation-dependent memory of somatic cell identity inhibits reprogramming and development of nuclear transfer embryos. Cell Stem Cell 21 , 135–143.e136 (2017).
Shimko, J. C. et al. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J. Mol. Biol. 408 , 187–204 (2011).
North, J. A. et al. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res . 40 , 10215–10227 (2012).
Tropberger, P. et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 152 , 859–872 (2013).
Spruce, C. et al. HELLS and PRDM9 form a pioneer complex to open chromatin at meiotic recombination hot spots. Genes Dev. 34 , 398–412 (2020).
Zhang, X., Liu, Y. & Wang, N. Multifaceted Roles of Histone Lysine Lactylation in Meiotic Gene Dynamics and Recombination. Preprint at https://www.biorxiv.org/content/10.1101/2024.01.25.576681v1 (2024).
Mihola, O. et al. Rat PRDM9 shapes recombination landscapes, duration of meiosis, gametogenesis, and age of fertility. BMC Biol. 19 , 86 (2021).
Kaiser, V. B. & Semple, C. A. Chromatin loop anchors are associated with genome instability in cancer and recombination hotspots in the germline. Genome Biol. 19 , 101 (2018).
Grey, C. et al. In vivo binding of PRDM9 reveals interactions with noncanonical genomic sites. Genome Res 27 , 580–590 (2017).
Liu, C., Zhang, Y., Liu, C. C. & Schatz, D. G. Structural insights into the evolution of the RAG recombinase. Nat. Rev. Immunol. 22 , 353–370 (2022).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100 , 57–70 (2000).
Arnould, C. et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590 , 660–665 (2021).
Thorslund, T. et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527 , 389–393 (2015).
Mattiroli, F. et al. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 150 , 1182–1195 (2012).
Pesavento, J. J., Yang, H., Kelleher, N. L. & Mizzen, C. A. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell Biol. 28 , 468–486, (2008).
Gong, F. et al. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell Biol. 216 , 1959–1974 (2017).
Li, X. et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. USA 111 , 7096–7101 (2014).
Yue, Q. et al. Histone H3K9 lactylation confers temozolomide resistance in glioblastoma via LUC7L2-mediated MLH1 intron retention. Adv. Sci. (Weinh.) 11 , e2309290 (2024).
PubMed Google Scholar
Kuo, A. J. et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484 , 115–119 (2012).
Beck, D. B. et al. The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes Dev. 26 , 2580–2589 (2012).
Rondinelli, B. et al. H3K4me3 demethylation by the histone demethylase KDM5C/JARID1C promotes DNA replication origin firing. Nucleic Acids Res 43 , 2560–2574 (2015).
Wu, R. et al. H3K9me3 demethylase Kdm4d facilitates the formation of pre-initiative complex and regulates DNA replication. Nucleic Acids Res 45 , 169–180 (2017).
Klein, K. N. et al. Replication timing maintains the global epigenetic state in human cells. Science 372 , 371–378 (2021).
Hong, H. et al. Global profiling of protein lysine lactylation and potential target modified protein analysis in hepatocellular carcinoma. Proteomics 23 , e2200432 (2023).
Huang, H. et al. p300-mediated lysine 2-hydroxyisobutyrylation regulates glycolysis. Mol. Cell 70 , 663–678.e666 (2018).
He, Y. et al. Numb/Parkin-directed mitochondrial fitness governs cancer cell fate via metabolic regulation of histone lactylation. Cell Rep. 42 , 112033 (2023).
Wang, Y. F. et al. Aspirin modulates succinylation of PGAM1K99 to restrict the glycolysis through NF-κB/HAT1/PGAM1 signaling in liver cancer. Acta Pharm. Sin. 44 , 211–220 (2023).
Pandkar, M. R., Sinha, S., Samaiya, A. & Shukla, S. Oncometabolite lactate enhances breast cancer progression by orchestrating histone lactylation-dependent c-Myc expression. Transl. Oncol. 37 , 101758 (2023).
Dmitrieva-Posocco, O. et al. β-Hydroxybutyrate suppresses colorectal cancer. Nature 605 , 160–165 (2022).
Wei, R. et al. Ketogenesis attenuates KLF5-dependent production of CXCL12 to overcome the immunosuppressive tumor microenvironment in colorectal cancer. Cancer Res 82 , 1575–1588 (2022).
Mehdikhani, F. et al. Histone butyrylation/ acetylation remains unchanged in triple negative breast cancer cells after a long term metabolic reprogramming. Asian Pac. J. Cancer Prev. 20 , 3597–3601 (2019).
Zhao, H. et al. Effect of ketogenic diets on body composition and metabolic parameters of cancer patients: A systematic review and meta-analysis. Nutrients 14 , 4192 (2022).
Jin, J., Byun, J. K., Choi, Y. K. & Park, K. G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 55 , 706–715 (2023).
Nie, L. B. et al. Global profiling of lysine 2-hydroxyisobutyrylome in Toxoplasma gondii using affinity purification mass spectrometry. Parasitol. Res. 119 , 4061–4071 (2020).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326 , 289–293 (2009).
Crane, E. et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523 , 240–244 (2015).
Feng, Y. et al. Simultaneous epigenetic perturbation and genome imaging reveal distinct roles of H3K9me3 in chromatin architecture and transcription. Genome Biol. 21 , 296 (2020).
Bian, Q., Anderson, E. C., Yang, Q. & Meyer, B. J. Histone H3K9 methylation promotes formation of genome compartments in Caenorhabditis elegans via chromosome compaction and perinuclear anchoring. Proc. Natl Acad. Sci. USA 117 , 11459–11470 (2020).
Montavon, T. et al. Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat. Commun. 12 , 4359 (2021).
Boettiger, A. N. et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529 , 418–422 (2016).
Feinberg, A. P., Ohlsson, R. & Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet 7 , 21–33 (2006).
Claes, B., Buysschaert, I. & Lambrechts, D. Pharmaco-epigenomics: Discovering therapeutic approaches and biomarkers for cancer therapy. Heredity (Edinb.) 105 , 152–160 (2010).
Camuzi, D. et al. Regulation is in the air: The relationship between hypoxia and epigenetics in cancer. Cells 8 , 300 (2019).
Yu, J. et al. Histone lactylation drives oncogenesis by facilitating m(6)A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 22 , 85 (2021).
Chen, X., Zhou, X. & Wang, X. m(6)A binding protein YTHDF2 in cancer. Exp. Hematol. Oncol. 11 , 21 (2022).
Xie, B. et al. CircXRN2 suppresses tumor progression driven by histone lactylation through activating the Hippo pathway in human bladder cancer. Mol. Cancer 22 , 151 (2023).
Huang, Z. W. et al. STAT5 promotes PD-L1 expression by facilitating histone lactylation to drive immunosuppression in acute myeloid leukemia. Signal Transduct. Target Ther. 8 , 391 (2023).
Yang, J. et al. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRβ signaling drives clear cell renal cell carcinoma progression. Int. J. Biol. Sci. 18 , 3470–3483 (2022).
Yue, B. et al. Linc00152 functions as a competing endogenous RNA to confer oxaliplatin resistance and holds prognostic values in colon cancer. Mol. Ther. 24 , 2064–2077 (2016).
Wang, J. et al. Enterobacterial LPS-inducible LINC00152 is regulated by histone lactylation and promotes cancer cells invasion and migration. Front Cell Infect. Microbiol 12 , 913815 (2022).
Qu, M. et al. Hypoxia Increases ATX expression by histone crotonylation in a HIF-2α-dependent manner. Int. J. Mol. Sci. 24 , 7031 (2023).
Liao, M. et al. Reduction of H3K27cr modification during DNA damage in colon cancer. Front Oncol. 12 , 924061 (2022).
Hou, J. Y. et al. Histone crotonylation of peripheral blood mononuclear cells is a potential biomarker for diagnosis of colorectal cancer. Epigenetics Chromatin 16 , 35 (2023).
Liu, N. et al. Histone H3 lysine 27 crotonylation mediates gene transcriptional repression in chromatin. Mol. Cell 83 , 2206–2221.e2211 (2023).
Lu, M. et al. Elevated histone H3 citrullination is associated with increased Beclin1 expression in HBV-related hepatocellular carcinoma. J. Med Virol. 92 , 1221–1230 (2020).
Yang, G. et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 22 , e50967 (2021).
Koronowski, K. B. et al. Ketogenesis impact on liver metabolism revealed by proteomics of lysine β-hydroxybutyrylation. Cell Rep. 36 , 109487 (2021).
Renehan, A. G. et al. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371 , 569–578 (2008).
Zhang, H. et al. MTA2 triggered R-loop trans-regulates BDH1-mediated β-hydroxybutyrylation and potentiates propagation of hepatocellular carcinoma stem cells. Signal Transduct. Target Ther. 6 , 135 (2021).
Zheng, Y. et al. PADI4 has genetic susceptibility to gastric carcinoma and upregulates CXCR2, KRT14 and TNF-α expression levels. Oncotarget 7 , 62159–62176 (2016).
Song, S. et al. A novel citrullinated modification of histone 3 and its regulatory mechanisms related to IPO-38 antibody-labeled protein. Front Oncol. 9 , 304 (2019).
Wang, L. et al. PADI2-mediated citrullination promotes prostate cancer progression. Cancer Res 77 , 5755–5768 (2017).
Cherrington, B. D. et al. Potential role for PAD2 in gene regulation in breast cancer cells. PLoS One 7 , e41242 (2012).
Xu, X. et al. The effects of histone crotonylation and bromodomain protein 4 on prostate cancer cell lines. Transl. Androl. Urol. 10 , 900–914 (2021).
Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 12 , 31–46 (2022).
Song, H. et al. Histone post-translational modification and the DNA damage response. Genes Dis. 10 , 1429–1444 (2023).
Lu, Y. et al. Global landscape of 2-hydroxyisobutyrylation in human pancreatic cancer. Front Oncol. 12 , 1001807 (2022).
Zhang, Z. et al. Lysine 2-hydroxyisobutyrylation proteomics reveals protein modification alteration in the actin cytoskeleton pathway of oral squamous cell carcinoma. J. Proteom. 249 , 104371 (2021).
Tanikawa, C. et al. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat. Commun. 3 , 676 (2012).
Mauracher, L. M. et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 16 , 508–518 (2018).
Hisada, Y. et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica 105 , 218–225 (2020).
Thålin, C. et al. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS One 13 , e0191231 (2018).
DeVore, S. B. et al. Histone citrullination represses MicroRNA expression, resulting in increased oncogene mRNAs in somatolactotrope cells. Mol. Cell Biol. 38 , e00084–18 (2018).
Song, J. et al. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. USA 101 , 14373–14378 (2004).
Li, H. et al. Ubc9 promotes invasion and metastasis of lung cancer cells. Oncol. Rep. 29 , 1588–1594 (2013).
Xiao, J. et al. UBC9 deficiency enhances immunostimulatory macrophage activation and subsequent antitumor T cell response in prostate cancer. J. Clin. Invest 133 , e158352 (2023).
Qin, Y. et al. BRCA1 proteins regulate growth of ovarian cancer cells by tethering Ubc9. Am. J. Cancer Res . 2 , 540–548 (2012).
Chen, C. et al. SUMOylation promotes extracellular vesicle-mediated transmission of lncRNA ELNAT1 and lymph node metastasis in bladder cancer. J. Clin. Invest 131 , e146431 (2021).
Moschos, S. J. et al. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: Identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene 26 , 4216–4225 (2007).
Poli, G. et al. Epigenetic mechanisms of inflammasome regulation. Int. J. Mol. Sci. 21 , 5758 (2020).
Cole, J., Morris, P., Dickman, M. J. & Dockrell, D. H. The therapeutic potential of epigenetic manipulation during infectious diseases. Pharm. Ther. 167 , 85–99 (2016).
Kulej, K. et al. Time-resolved global and chromatin proteomics during herpes simplex virus type 1 (HSV-1) infection. Mol. Cell Proteom. 16 , S92–s107 (2017).
Flecken, T. et al. Mapping the heterogeneity of histone modifications on Hepatitis B virus DNA using liver needle biopsies obtained from chronically infected patients. J. Virol. 93 , e02036–18 (2019).
Li, Z. et al. The KAT5-Acetyl-Histone4-Brd4 axis silences HIV-1 transcription and promotes viral latency. PLoS Pathog. 14 , e1007012 (2018).
Chu, X. et al. Lactylated histone H3K18 as a potential biomarker for the diagnosis and predicting the severity of septic shock. Front Immunol. 12 , 786666 (2021).
Soetkamp, D. et al. Myofilament phosphorylation in stem cell treated diastolic heart failure. Circ. Res. 129 , 1125–1140 (2021).
Nahrendorf, M. & Swirski, F. K. Innate immune cells in ischaemic heart disease: Does myocardial infarction beget myocardial infarction? Eur. Heart J. 37 , 868–872, (2016).
Dutta, P. et al. Myocardial infarction activates CCR2(+) hematopoietic stem and progenitor cells. Cell Stem Cell 16 , 477–487 (2015).
Manz, M. G. & Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 14 , 302–314 (2014).
Marinković, G. et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur. Heart J. 40 , 2713–2723 (2019).
Prabhu, S. D. & Frangogiannis, N. G. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res 119 , 91–112 (2016).
Wang, N. et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ. Res . 131 , 893–908 (2022).
Caudrillier, A. et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest 122 , 2661–2671 (2012).
Clark, S. R. et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med 13 , 463–469 (2007).
de Boer, O. J. et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 109 , 290–297 (2013).
Fuchs, T. A. et al. Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. USA 107 , 15880–15885 (2010).
Giles, J. T. et al. Myocardial citrullination in rheumatoid arthritis: A correlative histopathologic study. Arthritis Res Ther. 14 , R39 (2012).
Koushik, S. et al. PAD4: pathophysiology, current therapeutics and future perspective in rheumatoid arthritis. Expert Opin. Ther. Targets 21 , 433–447 (2017).
Ohlsson, S. M. et al. Neutrophils from vasculitis patients exhibit an increased propensity for activation by anti-neutrophil cytoplasmic antibodies. Clin. Exp. Immunol. 176 , 363–372 (2014).
Xu, J. et al. Extracellular histones are major mediators of death in sepsis. Nat. Med . 15 , 1318–1321 (2009).
Fert-Bober, J. et al. Citrullination of myofilament proteins in heart failure. Cardiovasc Res . 108 , 232–242 (2015).
Martinod, K. et al. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med . 214 , 439–458 (2017).
Meyer, A. et al. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 119 , 1064–1074 (2012).
Savchenko, A. S. et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 123 , 141–148 (2014).
Spencer, V. A. & Davie, J. R. Role of covalent modifications of histones in regulating gene expression. Gene 240 , 1–12 (1999).
Horckmans, M. et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 38 , 187–197 (2017).
Brill, A. et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 10 , 136–144 (2012).
Fuchs, T. A., Brill, A. & Wagner, D. D. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb. Vasc. Biol. 32 , 1777–1783, (2012).
Bandyopadhyay, G. K., Yu, J. G., Ofrecio, J. & Olefsky, J. M. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes 55 , 2277–2285, (2006).
Goudarzi, A. et al. Starvation promotes histone lysine butyrylation in the liver of male but not female mice. Gene 745 , 144647 (2020).
Zhang, Q. et al. Identification of histone malonylation in the human fetal brain and implications for diabetes-induced neural tube defects. Mol. Genet Genom. Med 8 , e1403 (2020).
Wu, J. et al. Endothelial cell-derived lactate triggers bone mesenchymal stem cell histone lactylation to attenuate osteoporosis. Adv. Sci. (Weinh.) 10 , e2301300 (2023).
Bhattacharya, S., Piya, S. & Borthakur, G. Bromodomain inhibitors: What does the future hold? Clin. Adv. Hematol. Oncol. 16 , 504–515 (2018).
Portela, A. & Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 28 , 1057–1068, (2010).
Urdinguio, R. G., Sanchez-Mut, J. V. & Esteller, M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 8 , 1056–1072, (2009).
Shukla, S. & Tekwani, B. L. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front Pharm. 11 , 537 (2020).
Gold, P. W. The organization of the stress system and its dysregulation in depressive illness. Mol. Psychiatry 20 , 32–47 (2015).
Gold, P. W. et al. Cardiac implications of increased arterial entry and reversible 24-h central and peripheral norepinephrine levels in melancholia. Proc. Natl Acad. Sci. USA 102 , 8303–8308 (2005).
Gold, P. W. et al. Responses to corticotropin-releasing hormone in the hypercortisolism of depression and Cushing’s disease. Pathophysiologic and diagnostic implications. N. Engl. J. Med 314 , 1329–1335 (1986).
Heinrich, P. C., Castell, J. V. & Andus, T. Interleukin-6 and the acute phase response. Biochem J. 265 , 621–636, (1990).
Duman, R. S., Heninger, G. R. & Nestler, E. J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54 , 597–606 (1997).
Pechtel, P. & Pizzagalli, D. A. Effects of early life stress on cognitive and affective function: an integrated review of human literature. Psychopharmacol. (Berl.) 214 , 55–70 (2011).
Bekhuis, E. et al. The network structure of major depressive disorder, generalized anxiety disorder and somatic symptomatology. Psychol. Med 46 , 2989–2998 (2016).
Ready, R. E., Mather, M. A., Santorelli, G. D. & Santospago, B. P. Apathy, alexithymia, and depressive symptoms: Points of convergence and divergence. Psychiatry Res 244 , 306–311, (2016).
Wu, M. S. et al. Effects of histone modification in major depressive disorder. Curr. Neuropharmacol. 20 , 1261–1277 (2022).
Yamanashi, T. et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci. Rep. 7 , 7677 (2017).
Kajitani, N. et al. Prefrontal cortex infusion of beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, produces antidepressant-like effects in a rodent model of depression. Neuropsychopharmacol. Rep. 40 , 157–165 (2020).
Pan, S. et al. Evaluation of the antidepressive property of β-hydroxybutyrate in mice. Behav. Pharm. 31 , 322–332 (2020).
Chen, L., Miao, Z. & Xu, X. β-hydroxybutyrate alleviates depressive behaviors in mice possibly by increasing the histone3-lysine9-β-hydroxybutyrylation. Biochem Biophys. Res Commun. 490 , 117–122 (2017).
Coryell, W., Mills, J., Dindo, L. & Calarge, C. A. Predictors of depressive symptom trajectories in a prospective follow-up of late adolescents. Psychol. Med 50 , 2283–2288 (2020).
Wang, Q. & Dwivedi, Y. Advances in novel molecular targets for antidepressants. Prog. Neuropsychopharmacol. Biol. Psychiatry 104 , 110041 (2021).
Moreno, C. L. & Mobbs, C. V. Epigenetic mechanisms underlying lifespan and age-related effects of dietary restriction and the ketogenic diet. Mol. Cell Endocrinol. 455 , 33–40 (2017).
Cui, H. et al. Lung myofibroblasts promote macrophage profibrotic activity through lactate-induced histone lactylation. Am. J. Respir. Cell Mol. Biol. 64 , 115–125 (2021).
Li, J. et al. Urban airborne PM(2.5) induces pulmonary fibrosis through triggering glycolysis and subsequent modification of histone lactylation in macrophages. Ecotoxicol. Environ. Saf. 273 , 116162 (2024).
Huang, S. et al. Quantitative proteomics analysis of lysine 2-hydroxyisobutyrylation in IgA nephropathy. Clin. Proteom. 18 , 7 (2021).
Chen, W. et al. Comprehensive analysis of lysine crotonylation in proteome of maintenance hemodialysis patients. Med. (Baltim.) 97 , e12035 (2018).
Hu, S. et al. Salvianolic acid B alleviates liver injury by regulating lactate-mediated histone lactylation in macrophages. Molecules 29 , 236 (2024).
Koguchi-Yoshioka, H. et al. Serum lactate dehydrogenase level as a possible predictor of treatment preference in psoriasis. J. Dermatol Sci. 103 , 109–115 (2021).
Takeo, N. et al. Hereditary lactate dehydrogenase M-subunit deficiency with late-developing pustular psoriasis-like lesions. J. Dermatol 43 , 1429–1432 (2016).
Achari, A. E. & Jain, S. K. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int. J. Mol. Sci. 18 , 1321 (2017).
da Silva Rosa, S. C., Liu, M. & Sweeney, G. Adiponectin synthesis, secretion and extravasation from circulation to interstitial space. Physiol. (Bethesda) 36 , 134–149 (2021).
Google Scholar
Ruiyang, B. et al. Adiponectin in psoriasis and its comorbidities: A review. Lipids Health Dis. 20 , 87 (2021).
Zhao, S., Wu, T., Fu, M. & Zhang, Z. Histone lactylation participates in psoriasis progression by regulating the adiponectin expression. Clin. Cosmet. Investig. Dermatol 17 , 219–227 (2024).
Yoshino, M. et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 372 , 1224–1229 (2021).
Yuan, H. et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 119 , 1904–1914 (2012).
Tervo, A. J. et al. An in silico approach to discovering novel inhibitors of human sirtuin type 2. J. Med. Chem. 47 , 6292–6298 (2004).
Taylor, D. M. et al. A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem. Biol. 6 , 540–546 (2011).
Chopra, V. et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2 , 1492–1497 (2012).
Bedalov, A. et al. Identification of a small molecule inhibitor of Sir2p. Proc. Natl Acad. Sci. USA 98 , 15113–15118 (2001).
Grozinger, C. M. et al. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 276 , 38837–38843 (2001).
Napper, A. D. et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 48 , 8045–8054 (2005).
Solomon, J. M. et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell Biol. 26 , 28–38 (2006).
Rumpf, T. et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 6 , 6263 (2015).
Lain, S. et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13 , 454–463 (2008).
Voogd, T. E., Vansterkenburg, E. L., Wilting, J. & Janssen, L. H. Recent research on the biological activity of suramin. Pharm. Rev. 45 , 177–203 (1993).
Zoltner, M. et al. Suramin exposure alters cellular metabolism and mitochondrial energy production in African trypanosomes. J. Biol. Chem. 295 , 8331–8347 (2020).
Kim, Y. Y. et al. AGK2 ameliorates mast cell-mediated allergic airway inflammation and fibrosis by inhibiting FcεRI/TGF-β signaling pathway. Pharm. Res 159 , 105027 (2020).
Tervo, A. J. et al. Discovering inhibitors of human sirtuin type 2: novel structural scaffolds. J. Med Chem. 49 , 7239–7241 (2006).
Wu, Q. J. et al. The sirtuin family in health and disease. Signal Transduct. Target Ther. 7 , 402 (2022).
Mondal, S. & Thompson, P. R. Protein arginine deiminases (PADs): Biochemistry and chemical biology of protein citrullination. Acc. Chem. Res 52 , 818–832 (2019).
Witalison, E. E., Thompson, P. R. & Hofseth, L. J. Protein arginine deiminases and associated citrullination: Physiological functions and diseases associated with dysregulation. Curr. Drug Targets 16 , 700–710 (2015).
Nakayama-Hamada, M. et al. Citrullinated fibrinogen inhibits thrombin-catalysed fibrin polymerization. J. Biochem 144 , 393–398 (2008).
Sipilä, K. H. et al. Extracellular citrullination inhibits the function of matrix associated TGF-β. Matrix Biol. 55 , 77–89 (2016).
Alghamdi, M. et al. An interplay of structure and intrinsic disorder in the functionality of peptidylarginine deiminases, a family of key autoimmunity-related enzymes. Cell Mol. Life Sci. 76 , 4635–4662 (2019).
Ying, S. et al. Transcriptional regulation of peptidylarginine deiminase expression in human keratinocytes. J. Dermatol Sci. 53 , 2–9 (2009).
Jang, B. et al. Subcellular localization of peptidylarginine deiminase 2 and citrullinated proteins in brains of scrapie-infected mice: nuclear localization of PAD2 and membrane fraction-enriched citrullinated proteins. J. Neuropathol. Exp. Neurol. 70 , 116–124 (2011).
Kan, R. et al. Potential role for PADI-mediated histone citrullination in preimplantation development. BMC Dev. Biol. 12 , 19 (2012).
Moelants, E.A.V. et al. Peptidylarginine deiminases: physiological function, interaction with chemokines and role in pathology. Drug Discov. Today Technol. 9 , e227–314 (2012).
Bicker, K. L. & Thompson, P. R. The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 99 , 155–163, (2013).
Causey, C. P. et al. The development of N-α-(2-carboxyl)benzoyl-N(5)-(2-fluoro-1-iminoethyl)-l-ornithine amide (o-F-amidine) and N-α-(2-carboxyl)benzoyl-N(5)-(2-chloro-1-iminoethyl)-l-ornithine amide (o-Cl-amidine) as second generation protein arginine deiminase (PAD) inhibitors. J. Med. Chem. 54 , 6919–6935 (2011).
Pritzker, L. B. & Moscarello, M. A. A novel microtubule independent effect of paclitaxel: the inhibition of peptidylarginine deiminase from bovine brain. Biochim. Biophys. Acta 1388 , 154–160, (1998).
Knuckley, B. et al. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry 49 , 4852–4863 (2010).
Knight, J. S. et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 114 , 947–956 (2014).
Wang, Y. et al. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J. Biol. Chem. 287 , 25941–25953 (2012).
Wang, Y. et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184 , 205–213 (2009).
Kolaczkowska, E. & Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13 , 159–175 (2013).
Al-U’datt, D. G. F. et al. Current knowledge into the role of the peptidylarginine deiminase (PAD) enzyme family in cardiovascular disease. Eur. J. Pharm. 891 , 173765 (2021).
Darrah, E., Rosen, A., Giles, J. T. & Andrade, F. Peptidylarginine deiminase 2, 3 and 4 have distinct specificities against cellular substrates: novel insights into autoantigen selection in rheumatoid arthritis. Ann. Rheum. Dis. 71 , 92–98, (2012).
Slack, J. L., Causey, C. P. & Thompson, P. R. Protein arginine deiminase 4: a target for an epigenetic cancer therapy. Cell Mol. Life Sci. 68 , 709–720, (2011).
Jones, J. E. et al. Synthesis and screening of a haloacetamidine containing library to identify PAD4 selective inhibitors. ACS Chem. Biol. 7 , 160–165 (2012).
Cantariño, N. et al. Downregulation of the deiminase PADI2 is an early event in colorectal carcinogenesis and indicates poor prognosis. Mol. Cancer Res 14 , 841–848 (2016).
Knuckley, B. et al. Haloacetamidine-based inactivators of protein arginine deiminase 4 (PAD4): evidence that general acid catalysis promotes efficient inactivation. Chembiochem 11 , 161–165 (2010).
Teo, C. Y. et al. Novel furan-containing peptide-based inhibitors of protein arginine deiminase type IV (PAD4). Chem. Biol. Drug Des. 90 , 1134–1146 (2017).
Witalison, E. E. et al. Molecular targeting of protein arginine deiminases to suppress colitis and prevent colon cancer. Oncotarget 6 , 36053–36062 (2015).
Demers, M. & Wagner, D. D. NETosis: a new factor in tumor progression and cancer-associated thrombosis. Semin Thromb. Hemost. 40 , 277–283, (2014).
McNee, G. et al. Citrullination of histone H3 drives IL-6 production by bone marrow mesenchymal stem cells in MGUS and multiple myeloma. Leukemia 31 , 373–381 (2017).
Yao, H. et al. Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J. Biol. Chem. 283 , 20060–20068 (2008).
Cedervall, J., Zhang, Y. & Olsson, A. K. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res 76 , 4311–4315, (2016).
Tao, L. et al. Polypharmacological profiles underlying the antitumor property of salvia miltiorrhiza root (Danshen) interfering with NOX-dependent neutrophil extracellular traps. Oxid. Med. Cell Longev. 2018 , 4908328 (2018).
Zhu, D. et al. Highly-tumor-targeted PAD4 inhibitors with PBA modification inhibit tumors in vivo by specifically inhibiting the PAD4-H3cit-NETs pathway in neutrophils. Eur. J. Med Chem. 258 , 115619 (2023).
Yu, Y., Huang, X., Liang, C. & Zhang, P. Evodiamine impairs HIF1A histone lactylation to inhibit Sema3A-mediated angiogenesis and PD-L1 by inducing ferroptosis in prostate cancer. Eur. J. Pharm. 957 , 176007 (2023).
Li, M. et al. A novel peptidylarginine deiminase 4 (PAD4) inhibitor BMS-P5 blocks formation of neutrophil extracellular traps and delays progression of multiple myeloma. Mol. Cancer Ther. 19 , 1530–1538 (2020).
Tejeda, E. J. C. et al. Noncovalent protein arginine deiminase (PAD) inhibitors are efficacious in animal models of multiple sclerosis. J. Med Chem. 60 , 8876–8887 (2017).
Yao, H. et al. Inhibition of netosis with PAD inhibitor attenuates endotoxin shock induced systemic inflammation. Int. J. Mol. Sci. 23 , 13264 (2022).
Dokmanovic, M. & Marks, P. A. Prospects: histone deacetylase inhibitors. J. Cell Biochem 96 , 293–304 (2005).
Dokmanovic, M., Clarke, C. & Marks, P. A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res 5 , 981–989, (2007).
Weaver, I. C., Meaney, M. J. & Szyf, M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc. Natl Acad. Sci. USA 103 , 3480–3485, (2006).
Kv, A. et al. Antidepressant activity of vorinostat is associated with amelioration of oxidative stress and inflammation in a corticosterone-induced chronic stress model in mice. Behav. Brain Res. 344 , 73–84 (2018).
Meylan, E. M., Halfon, O., Magistretti, P. J. & Cardinaux, J. R. The HDAC inhibitor SAHA improves depressive-like behavior of CRTC1-deficient mice: Possible relevance for treatment-resistant depression. Neuropharmacology 107 , 111–121 (2016).
Calabrese, F. et al. Modulation of neuronal plasticity following chronic concomitant administration of the novel antipsychotic lurasidone with the mood stabilizer valproic acid. Psychopharmacol. (Berl.) 226 , 101–112 (2013).
Wu, H. F. et al. Alleviation of N-Methyl-D-aspartate receptor-dependent long-term depression via regulation of the glycogen synthase kinase-3β pathway in the amygdala of a valproic acid-induced animal model of autism. Mol. Neurobiol. 54 , 5264–5276 (2017).
Goudarzi, M. et al. Valproic acid administration exerts protective effects against stress-related anhedonia in rats. J. Chem. Neuroanat. 105 , 101768 (2020).
Lin, H. et al. Molecular mechanisms associated with the antidepressant effects of the class I histone deacetylase inhibitor MS-275 in the rat ventrolateral orbital cortex. Brain Res . 1447 , 119–125 (2012).
Hubbert, C. et al. HDAC6 is a microtubule-associated deacetylase. Nature 417 , 455–458 (2002).
Jeong, J. W. et al. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 111 , 709–720 (2002).
Yuan, Z. L., Guan, Y. J., Chatterjee, D. & Chin, Y. E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307 , 269–273, (2005).
Wolf, D. et al. Acetylation of beta-catenin by CREB-binding protein (CBP). J. Biol. Chem. 277 , 25562–25567 (2002).
Laubach, J. P. et al. Efficacy and safety of oral panobinostat plus subcutaneous bortezomib and oral dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma (PANORAMA 3): an open-label, randomised, phase 2 study. Lancet Oncol. 22 , 142–154 (2021).
Ookubo, M., Kanai, H., Aoki, H. & Yamada, N. Antidepressants and mood stabilizers effects on histone deacetylase expression in C57BL/6 mice: Brain region specific changes. J. Psychiatr. Res 47 , 1204–1214, (2013).
Wu, S. et al. Lithium down-regulates histone deacetylase 1 (HDAC1) and induces degradation of mutant huntingtin. J. Biol. Chem. 288 , 35500–35510 (2013).
Seo, M. K. et al. Effects of antipsychotic drugs on the epigenetic modification of brain-derived neurotrophic factor gene expression in the hippocampi of chronic restraint stress rats. Neural Plast. 2018 , 2682037 (2018).
Yasuda, S. et al. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 14 , 51–59 (2009).
Leu, S. J. et al. Valproic acid and lithium meditate anti-inflammatory effects by differentially modulating dendritic cell differentiation and function. J. Cell Physiol. 232 , 1176–1186 (2017).
Feng, D. D. et al. Nine traditional Chinese herbal formulas for the treatment of depression: An ethnopharmacology, phytochemistry, and pharmacology review. Neuropsychiatr. Dis. Treat. 12 , 2387–2402 (2016).
Aggarwal, A. et al. Quercetin alleviates cognitive decline in ovariectomized mice by potentially modulating histone acetylation homeostasis. J. Nutr. Biochem 84 , 108439 (2020).
Chen, P. S. et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatry 11 , 1116–1125 (2006).
Wu, X. et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J. Neuropsychopharmacol. 11 , 1123–1134, (2008).
Sharma, S., Taliyan, R. & Singh, S. Beneficial effects of sodium butyrate in 6-OHDA induced neurotoxicity and behavioral abnormalities: Modulation of histone deacetylase activity. Behav. Brain Res 291 , 306–314 (2015).
Chen, S. H. et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Br. J. Pharm. 165 , 494–505 (2012).
Kontopoulos, E., Parvin, J. D. & Feany, M. B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet 15 , 3012–3023, (2006).
Nicholas, A. P. et al. Striatal histone modifications in models of levodopa-induced dyskinesia. J. Neurochem 106 , 486–494 (2008).
Su, Y. et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 43 , 6899–6908 (2004).
Qing, H. et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J. Exp. Med 205 , 2781–2789 (2008).
Zhang, L. et al. Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 41 , 1193–1205 (2014).
Jian, W. X. et al. Donepezil attenuates vascular dementia in rats through increasing BDNF induced by reducing HDAC6 nuclear translocation. Acta Pharm. Sin. 41 , 588–598 (2020).
Dompierre, J. P. et al. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 27 , 3571–3583 (2007).
Ferrante, R. J. et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 23 , 9418–9427 (2003).
Jia, H. et al. The effects of pharmacological inhibition of histone deacetylase 3 (HDAC3) in Huntington’s disease mice. PLoS One 11 , e0152498 (2016).
Chopra, V. et al. LBH589, A hydroxamic acid-derived HDAC inhibitor, is neuroprotective in mouse models of Huntington’s disease. J. Huntingt. Dis. 5 , 347–355 (2016).
Hahnen, E. et al. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J. Neurochem 98 , 193–202 (2006).
Tsai, L. K., Yang, C. C., Hwu, W. L. & Li, H. Valproic acid treatment in six patients with spinal muscular atrophy. Eur. J. Neurol. 14 , e8–e9 (2007).
Minamiyama, M. et al. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet 13 , 1183–1192 (2004).
Liu, H. et al. The Smn-independent beneficial effects of trichostatin A on an intermediate mouse model of spinal muscular atrophy. PLoS One 9 , e101225 (2014).
Hauke, J. et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum. Mol. Genet 18 , 304–317 (2009).
Brahe, C. et al. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur. J. Hum. Genet 13 , 256–259 (2005).
Jiang, C. et al. The potential mechanism of HDAC1-catalyzed histone crotonylation of caspase-1 in nonsmall cell lung cancer. Evid. Based Complement Altern. Med 2022 , 5049116 (2022).
Singh, B., Boopathy, S., Somasundaram, K. & Umapathy, S. Fourier transform infrared microspectroscopy identifies protein propionylation in histone deacetylase inhibitor treated glioma cells. J. Biophotonics 5 , 230–239 (2012).
Xu, G. et al. SAHA regulates histone acetylation, butyrylation, and protein expression in neuroblastoma. J. Proteome Res . 13 , 4211–4219 (2014).
Yuan, Y. et al. Aspirin modulates 2-hydroxyisobutyrylation of ENO1K281 to attenuate the glycolysis and proliferation of hepatoma cells. Biochem Biophys. Res Commun. 560 , 172–178 (2021).
Lee, S. Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 3 , 198–210 (2016).
Zhang, B. et al. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 27 , 45–52 (2022).
Chen, J. et al. Histone lactylation driven by mROS-mediated glycolytic shift promotes hypoxic pulmonary hypertension. J. Mol. Cell Biol. 14 , mjac073 (2023).
Li, W. et al. Tumor-derived lactate promotes resistance to bevacizumab treatment by facilitating autophagy enhancer protein RUBCNL expression through histone H3 lysine 18 lactylation (H3K18la) in colorectal cancer. Autophagy 20 , 114–130 (2024).
Rho, H., Terry, A. R., Chronis, C. & Hay, N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab. 35 , 1406–1423.e8 (2023).
Li, X. M. et al. Histone lactylation inhibits RARγ expression in macrophages to promote colorectal tumorigenesis through activation of TRAF6-IL-6-STAT3 signaling. Cell Rep. 43 , 113688 (2024).
Zhao, Y. et al. Comprehensive analysis for histone acetylation of human colon cancer cells treated with a novel HDAC inhibitor. Curr. Pharm. Des. 20 , 1866–1873 (2014).
Hegedüs, R. et al. Enhanced cellular uptake and in vitro antitumor activity of short-chain fatty acid acylated daunorubicin-GnRH-III bioconjugates. Eur. J. Med Chem. 56 , 155–165 (2012).
Kapuvári, B. et al. Improved in vivo antitumor effect of a daunorubicin - GnRH-III bioconjugate modified by apoptosis inducing agent butyric acid on colorectal carcinoma bearing mice. Invest N. Drugs 34 , 416–423 (2016).
Pan, L. et al. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharm. Res 181 , 106270 (2022).
Xu, H. et al. Royal jelly acid suppresses hepatocellular carcinoma tumorigenicity by inhibiting H3 histone lactylation at H3K9la and H3K14la sites. Phytomedicine 118 , 154940 (2023).
Yuan, H. et al. Lysine catabolism reprograms tumour immunity through histone crotonylation. Nature 617 , 818–826 (2023).
Wang, C. et al. Andrographolide regulates H3 histone lactylation by interfering with p300 to alleviate aortic valve calcification. Br. J. Pharmacol , (2024).
Nakato, R. & Shirahige, K. Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Brief. Bioinform 18 , 279–290 (2017).
Furey, T. S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet 13 , 840–852, (2012).
Liu, Y. et al. A practical guide for DNase-seq data analysis: from data management to common applications. Brief. Bioinform 20 , 1865–1877 (2019).
Yan, F., Powell, D. R., Curtis, D. J. & Wong, N. C. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 21 , 22 (2020).
Shen, Y., Chen, L. L. & Gao, J. CharPlant: A De Novo Open Chromatin Region Prediction Tool for Plant Genomes. Genomics Proteom. Bioinforma. 19 , 860–871 (2021).
Chereji, R. V., Bryson, T. D. & Henikoff, S. Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol. 20 , 198 (2019).
Farrell, C. et al. BiSulfite Bolt: A bisulfite sequencing analysis platform. Gigascience 10 , giab033 (2021).
Booth, M. J. et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336 , 934–937 (2012).
Xu, C. & Corces, V. G. Resolution of the DNA methylation state of single CpG dyads using in silico strand annealing and WGBS data. Nat. Protoc. 14 , 202–216 (2019).
Lu, X. et al. Chemical modification-assisted bisulfite sequencing (CAB-Seq) for 5-carboxylcytosine detection in DNA. J. Am. Chem. Soc. 135 , 9315–9317 (2013).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10 , 1930 (2019).
Bartosovic, M., Kabbe, M., Castelo-Branco, G. & Single-cell, C. U. T. &Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39 , 825–835 (2021).
Skene, P. J. & Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6 , e21856 (2017).
Chenarani, N. et al. Bioinformatic tools for DNA methylation and histone modification: A survey. Genomics 113 , 1098–1113 (2021).
Kwon, M. J., Kim, S., Han, M. H. & Lee, S. B. Epigenetic changes in neurodegenerative diseases. Mol. Cells 39 , 783–789 (2016).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129 , 823–837 (2007).
Skene, P. J., Henikoff, J. G. & Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13 , 1006–1019 (2018).
Yashar, W. M. et al. GoPeaks: histone modification peak calling for CUT&Tag. Genome Biol. 23 , 144 (2022).
Li, R., Grimm, S. A. & Wade, P. A. CUT&Tag-BS for simultaneous profiling of histone modification and DNA methylation with high efficiency and low cost. Cell Rep. Methods 1 , 100118 (2021).
Carter, B. et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun. 10 , 3747 (2019).
Yeung, J. et al. scChIX-seq infers dynamic relationships between histone modifications in single cells. Nat. Biotechnol. 41 , 813–823 (2023).
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D. & Thermes, C. The third revolution in sequencing technology. Trends Genet 34 , 666–681 (2018).
Meslier, V. et al. Benchmarking second and third-generation sequencing platforms for microbial metagenomics. Sci. Data 9 , 694 (2022).
Yang, S. et al. Whole genome assembly of human papillomavirus by nanopore long-read sequencing. Front Genet 12 , 798608 (2021).
Lu, H., Giordano, F. & Ning, Z. Oxford Nanopore MinION sequencing and genome assembly. Genomics Proteom. Bioinforma. 14 , 265–279 (2016).
Larkin, J. et al. Length-independent DNA packing into nanopore zero-mode waveguides for low-input DNA sequencing. Nat. Nanotechnol. 12 , 1169–1175 (2017).
Athanasopoulou, K. et al. Third-generation sequencing: The spearhead towards the radical transformation of modern genomics. Life (Basel) 12 , 30 (2021).
Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genomics Proteom. Bioinforma. 13 , 278–289 (2015).
Yue, X. et al. Simultaneous profiling of histone modifications and DNA methylation via nanopore sequencing. Nat. Commun. 13 , 7939 (2022).
Fan, X. et al. SMOOTH-seq: single-cell genome sequencing of human cells on a third-generation sequencing platform. Genome Biol. 22 , 195 (2021).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36 , 338–345 (2018).
Fenn, J. B. et al. Electrospray ionization for mass spectrometry of large biomolecules. Science 246 , 64–71 (1989).
Hillenkamp, F. & Karas, M. Mass spectrometry of peptides and proteins by matrix-assisted ultraviolet laser desorption/ionization. Methods Enzymol. 193 , 280–295, (1990).
Noberini, R. & Bonaldi, T. Mass spectrometry-based analysis of histone posttranslational modifications from laser microdissected samples. Methods Mol. Biol. 2718 , 271–283 (2023).
Karch, K. R., Sidoli, S. & Garcia, B. A. Identification and quantification of histone PTMs using high-resolution mass spectrometry. Methods Enzymol. 574 , 3–29 (2016).
Geffen, Y. et al. Pan-cancer analysis of post-translational modifications reveals shared patterns of protein regulation. Cell 186 , 3945–3967.e3926 (2023).
Aebersold, R., Burlingame, A. L. & Bradshaw, R. A. Western blots versus selected reaction monitoring assays: Time to turn the tables? Mol. Cell Proteom. 12 , 2381–2382, (2013).
Wang, H. et al. The clinical impact of recent advances in LC-MS for cancer biomarker discovery and verification. Expert Rev. Proteom. 13 , 99–114 (2016).
Noberini, R. et al. PAT-H-MS coupled with laser microdissection to study histone post-translational modifications in selected cell populations from pathology samples. Clin. Epigenetics 9 , 69 (2017).
Noberini, R. et al. Pathology tissue-quantitative mass spectrometry analysis to profile histone post-translational modification patterns in patient samples. Mol. Cell Proteom. 15 , 866–877 (2016).
Kriegsmann, J., Kriegsmann, M. & Casadonte, R. MALDI TOF imaging mass spectrometry in clinical pathology: a valuable tool for cancer diagnostics (review). Int. J. Oncol. 46 , 893–906 (2015).
Poté, N. et al. Imaging mass spectrometry reveals modified forms of histone H4 as new biomarkers of microvascular invasion in hepatocellular carcinomas. Hepatology 58 , 983–994 (2013).
Robusti, G., Vai, A., Bonaldi, T. & Noberini, R. Investigating pathological epigenetic aberrations by epi-proteomics. Clin. Epigenetics 14 , 145 (2022).
Sidoli, S. et al. One minute analysis of 200 histone posttranslational modifications by direct injection mass spectrometry. Genome Res 29 , 978–987 (2019).
Kelly, R. T. Single-cell proteomics: Progress and prospects. Mol. Cell Proteom. 19 , 1739–1748 (2020).
Graham, K. A., Lawlor, C. F. & Borotto, N. B. Characterizing the top-down sequencing of protein ions prior to mobility separation in a timsTOF. Analyst 148 , 1534–1542 (2023).
Zucker, S. M. et al. An ion mobility/ion trap/photodissociation instrument for characterization of ion structure. J. Am. Soc. Mass Spectrom. 22 , 1477–1485 (2011).
Miller, S. A. et al. Trapped ion mobility spectrometry, ultraviolet photodissociation, and time-of-flight mass spectrometry for gas-phase peptide isobars/isomers/conformers discrimination. J. Am. Soc. Mass Spectrom. 33 , 1267–1275 (2022).
Hawkes, J. A. et al. Evaluation of the orbitrap mass spectrometer for the molecular fingerprinting analysis of natural dissolved organic matter. Anal. Chem. 88 , 7698–7704 (2016).
Nicolardi, S. et al. Developments in FTICR-MS and its potential for body fluid signatures. Int. J. Mol. Sci. 16 , 27133–27144 (2015).
Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196 , 801–810 (2012).
Lambert, J. P. et al. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteom. 118 , 81–94 (2015).
Remnant, L. et al. In vitro BioID: Mapping the CENP-A microenvironment with high temporal and spatial resolution. Mol. Biol. Cell 30 , 1314–1325 (2019).
Goudarzi, A. et al. Dynamic competing histone H4 K5K8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol. Cell 62 , 169–180 (2016).
Nicolas, E., Roumillac, C. & Trouche, D. Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Mol. Cell Biol. 23 , 1614–1622, (2003).
Cedar, H. & Bergman, Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet 10 , 295–304 (2009).
Fuino, L. et al. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol. Cancer Ther. 2 , 971–984 (2003).
Yu, C. et al. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin. Cancer Res. 13 , 1140–1148 (2007).
Qian, D. Z. et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res 64 , 6626–6634 (2004).
Dai, S. et al. Fibroblast growth factor receptors (FGFRs): Structures and small molecule inhibitors. Cells 8 , 614 (2019).
Liu, J. et al. Design, synthesis and evaluate of novel dual FGFR1 and HDAC inhibitors bearing an indazole scaffold. Bioorg. Med Chem. 26 , 747–757 (2018).
Biersack, B., Polat, S. & Höpfner, M. Anticancer properties of chimeric HDAC and kinase inhibitors. Semin Cancer Biol. 83 , 472–486 (2022).
Abu-Zhayia, E. R., Machour, F. E. & Ayoub, N. HDAC-dependent decrease in histone crotonylation during DNA damage. J. Mol. Cell Biol. 11 , 804–806 (2019).
Song, X. et al. Dynamic crotonylation of EB1 by TIP60 ensures accurate spindle positioning in mitosis. Nat. Chem. Biol. 17 , 1314–1323 (2021).
Li, L. et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat. Metab. 2 , 882–892 (2020).
Irizarry-Caro, R. A. et al. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc. Natl Acad. Sci. USA 117 , 30628–30638 (2020).
Yi, L. et al. New mechanisms: From lactate to lactylation to rescue heart failure. Biosci. Trends 18 , 105–107 (2024).
Smestad, J., Erber, L., Chen, Y. & Maher, L. J. 3rd Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. iScience 2 , 63–75 (2018).
Wang, G. et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol. Cell 74 , 844–857.e847 (2019).
Subasinghe, S. et al. Cholesterol is necessary both for the toxic effect of Abeta peptides on vascular smooth muscle cells and for Abeta binding to vascular smooth muscle cell membranes. J. Neurochem 84 , 471–479 (2003).
Zhou, B. et al. Identification of malonylation, succinylation, and glutarylation in serum proteins of acute myocardial infarction patients. Proteom. Clin. Appl 14 , e1900103 (2020).
Es-Haghi, A., Shariatizi, S., Ebrahim-Habibi, A. & Nemat-Gorgani, M. Amyloid fibrillation in native and chemically-modified forms of carbonic anhydrase II: role of surface hydrophobicity. Biochim Biophys. Acta 1824 , 468–477, (2012).
Liu, S. et al. Genome-wide profiling of histone lysine butyrylation reveals its role in the positive regulation of gene transcription in rice. Rice (N. Y) 12 , 86 (2019).
Zhang, H. et al. Ketogenesis-generated β-hydroxybutyrate is an epigenetic regulator of CD8(+) T-cell memory development. Nat. Cell Biol. 22 , 18–25 (2020).
Luo, W. et al. Up-regulation of MMP-2 by histone H3K9 β-hydroxybutyrylation to antagonize glomerulosclerosis in diabetic rat. Acta Diabetol. 57 , 1501–1509 (2020).
Zhang, L. et al. The m6A reader YTHDF2 promotes bladder cancer progression by suppressing RIG-I-mediated immune response. Cancer Res 83 , 1834–1850 (2023).
Chaudagar, K. et al. Suppression of tumor cell lactate-generating signaling pathways eradicates murine PTEN/p53-deficient aggressive-variant prostate cancer via macrophage phagocytosis. Clin. Cancer Res . 29 , 4930–4940 (2023).
Wu, Q. et al. Integrated analysis of histone lysine lactylation (Kla)-specific genes suggests that NR6A1, OSBP2 and UNC119B are novel therapeutic targets for hepatocellular carcinoma. Sci. Rep. 13 , 18642 (2023).
Liao, J. et al. CENPA functions as a transcriptional regulator to promote hepatocellular carcinoma progression via cooperating with YY1. Int. J. Biol. Sci. 19 , 5218–5232 (2023).
Jin, J. et al. SIRT3-dependent delactylation of cyclin E2 prevents hepatocellular carcinoma growth. EMBO Rep. 24 , e56052 (2023).
Murata, T. et al. Transcriptional repression by sumoylation of Epstein-Barr virus BZLF1 protein correlates with association of histone deacetylase. J. Biol. Chem. 285 , 23925–23935 (2010).
Download references
Acknowledgements
This study was funded by National Natural Science Foundation (No.82270200, No.82070203 and No.81770210); Taishan Scholars Program of Shandong Province; Shandong Provincial Engineering Research Center of Lymphoma; Key Research and Development Program of Shandong Province (No.2018CXGC1213); Academic Promotion Programme of Shandong First Medical University (No. 2019QL018); Translational Research Grant of NCRCH (No.2021WWB02, No.2020ZKMB01).
Author information
Authors and affiliations.
Department of Hematology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, 250021, China
Weiyi Yao, Xinting Hu & Xin Wang
Department of Hematology, Shandong Provincial Hospital, Shandong University, Jinan, Shandong, 250021, China
Xinting Hu & Xin Wang
Taishan Scholars Program of Shandong Province, Jinan, Shandong, 250021, China
You can also search for this author in PubMed Google Scholar
Contributions
WY, XH, XW, wrote the paper; XH, XW, edited the paper; XW, final edits and submission. The authors read and approved the final manuscript. WY created the figures by Figdraw. All authors have read and approved the article.
Corresponding authors
Correspondence to Xinting Hu or Xin Wang .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Yao, W., Hu, X. & Wang, X. Crossing epigenetic frontiers: the intersection of novel histone modifications and diseases. Sig Transduct Target Ther 9 , 232 (2024). https://doi.org/10.1038/s41392-024-01918-w
Download citation
Received : 28 February 2024
Revised : 11 June 2024
Accepted : 30 June 2024
Published : 16 September 2024
DOI : https://doi.org/10.1038/s41392-024-01918-w
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies

IMAGES
VIDEO
COMMENTS
This points to a possible new era of infectious disease, defined by outbreaks of emerging, re-emerging and endemic pathogens that spread quickly, aided by global connectivity and shifted ranges ...
To adequately prepare for potential hazards caused by emerging and reemerging infectious diseases, the WHO has issued a list of high-priority pathogens that are likely to cause future outbreaks ...
Emerging Infectious Diseases is a peer-reviewed, monthly journal published by the Centers for Disease Control and Prevention (CDC). It offers global health professionals the latest scientific information on emerging infectious diseases and trends. Articles provide the most up-to-date information on infectious diseases and their effects on global health.
Definitions. 'Emerging infectious diseases' are defined as 'those whose incidence in humans has increased within the past two decades or threatens to increase in the near future'. Emergence can be from the spread of new agents (including known agents with a new resistance mechanism), from recognition of an infection that has been ...
Emerging infectious diseases (EIDs) and re-emerging infectious diseases (REIDs) are responsible for a significant proportion of the infectious disease outbreaks that have plagued humanity over the ages. EIDs are infectious diseases that have not occurred in humans before, have occurred previously in humans but affected only small populations in ...
As he prepares to step down from the NIAID after 54 years, Dr. Anthony Fauci reflects on the evolution of his field and cautions that emerging infectious diseases are truly a perpetual challenge.
This points to a possible new era of infectious disease, defined by outbreaks of emerging, re-emerging and endemic pathogens that spread quickly, aided by global connectivity and shifted ranges owing to climate change (Fig. 1d). Human connectivity and infectious disease outbreaks in premodern and modern times.
Main. Emerging infectious diseases are on the rise, often originate from wildlife, and are significantly correlated with socioeconomic, environmental and ecological factors 1. As a consequence ...
Page reviewed: August 22, 2024. The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions.
Emerging Infectious Diseases is a peer-reviewed, monthly journal published by the Centers for Disease Control and Prevention (CDC). It offers global health professionals the latest scientific information on emerging infectious diseases and trends. Articles provide the most up-to-date information on infectious diseases and their effects on global health.
Remarkable progress has been made in preventing deaths from infectious diseases. Now, attention could shift to focusing more resources on pandemic preparedness, including detecting and containing e...
Notably, 60 to 80 percent of new human infections likely originated in animals, disproportionately rodents and bats, as shown by the examples of hantavirus pulmonary syndrome, Lassa fever, and Nipah virus encephalitis [2] - [4]. Most other emerging/reemerging diseases result from human-adapted infectious agents that genetically acquire ...
Infectious diseases prevalent in humans and animals are caused by pathogens that once emerged from other animal hosts. In addition to these established infections, new infectious diseases periodically emerge. In extreme cases they may cause pandemics such as COVID-19; in other cases, dead-end infections or smaller epidemics result.
The unprecedented scale and rapidity of dissemination of recent emerging infectious diseases pose new challenges for vaccine developers, regulators, health authorities and political constituencies.
Vector-borne emerging and re-emerging diseases pose considerable public health problem worldwide. Some of these diseases are emerging and/or re-emerging at increasing rates and appeared in new regions in the past two decades. Studies emphasized that the ...
Emerging Infectious Diseases is a peer-reviewed, monthly journal published by the Centers for Disease Control and Prevention (CDC). It offers global health professionals the latest scientific information on emerging infectious diseases and trends. Articles provide the most up-to-date information on infectious diseases and their effects on global health.
This includes surveillance of circulating strains, research on the molecular properties of emerging virus groups, the administration of vaccines or antiviral drugs, as well as other disease-specific measures, which have been undertaken to help combat the vector-borne flavivirus disease burden [35-37].
Fig. 1: The global extent of newly emerging, re-emerging, and "deliberately emerging" infectious diseases from 1981 to the present (2020).
A synthesis work of empirical climate change and infectious disease research is still lacking. We conducted a systemic review of research from 2015 to 2020 period on climate change and infectious diseases to identify major trends and current gaps of research.
Emerging Infectious Diseases. Emerging infectious diseases (EID) and reemerging infectious diseases are increasing globally. Zoonotic diseases are transmitted from animals to humans through direct contact or through food, water, and the environment. Vector-borne diseases are major sources of mortality and morbidity globally.
Hepatitis E virus (HEV) is an interesting topic in the field of emerging infectious diseases. HEV ranks 6th on a list of spillover viruses with significant health risks to humans [].HEV causes both acute and chronic infection in humans and is the leading cause of acute viral gastroenteritis worldwide [].Chronic HEV infection has been reported in patients receiving a solid organ transplant (SOT ...
The twenty-first century has already recorded more than ten major epidemic or pandemic virus emergence events, including the ongoing and devastating coronavirus disease 2019 (COVID-19) pandemic ...
Dr. Davis is a postdoctoral fellow at the Arboviral Diseases Branch, Division of Vector-Borne Diseases, National Center for Emerging and Zoonotic Infectious Diseases, CDC. Her primary research interests include viral pathogenicity and genomics. Mr. Velez oversees the Arboviral Diseases Branch cell culture laboratory.
This review highlights some of the recent concerning emerging infectious diseases, a number of them specifically that the World Health Organization has categorized as priorities for research.Emerging and reemerging infectious diseases account for significant ...
The origins of emerging infections diseases are significantly correlated with socio-economic, environmental and ecological factors.
After decades of relative obscurity in the mid-20th century, infectious disease epidemiology has experienced an intellectual rebirth in response to disease emergence. Repopulation of this field by scientists trained not only in clinical medicine but ecology, demography, and quantitative sciences has led to the adoption of methods scarcely ...
Furthermore, a growing body of research has demonstrated that novel histone modifications also play crucial roles in the development and progression of various diseases, including various cancers ...